The first complete chloroplast genome of Polygonatum hirtellum: Genome features and phylogenetic relationships
Article information

Abstract
Polygonatum hirtellum is a perennial herb within the family Asparagaceae. In the present study, the chloroplast (cp) genome of P. hirtellum is reported for the first time, and its phylogenetic relationships are also investigated. The findings revealed that the cp genome was 155,697 bp and exhibited a typical quadripartite structure, consisting of a large single copy (84,468 bp), a small single copy (18,491 bp), and a pair of invert repeat sequences (26,369 bp). The GC content of the cp genomes amounted to 37.7%; 130 genes were identified, consisting of 84 protein-coding genes, 38 transfer RNA genes, and eight ribosomal RNA genes. A phylogenetic analysis based on the cp genome and coding sequence showed that P. hirtellum was a member of section Verticillata and could be distinguished from other species of the genus Polygonatum used in the analysis.
INTRODUCTION
The genus Polygonatum is a diverse plant group widely distributed in the north temperate zone, with about 75 species recognized worldwide (Chen and Tamura, 2000; Floden and Schilling, 2018). Many species of this genus possess extensive pharmacological activities, such as anti-tumor, immunomodulatory, and hypoglycemic activities (Chen et al., 2020, 2023; Wang et al., 2020). However, the wild resources of the genus Polygonatum have been devastated due to long-term predatory mining (Li et al., 2023). Strengthening the protection of germplasm resources and conducting related basic research on this genus of plants are urgent tasks. P. hirtellum, a rare species within the genus Polygonatum, grows at altitudes ranging from 1,000 to 2,900 m and has potential applications in the field of pharmacology (Yang, 2016; Xu et al., 2018). To date, the studies of P. hirtellum have mainly focused on morphological identification, pharmacological activities (Yang, 2016; Peng, 2018; Zhao et al., 2023), and chromosome ploidy (Deng et al., 2009). In terms of genetic information, Meng et al. (2014) reported several gene fragments, including rbcL, trnK, psbA-trnH, and trnC-petN (Meng et al., 2014). However, the genetic information of P. hirtellum has not yet been fully revealed, limiting our understanding of its genetic background, related germplasm resources, and phylogenetic evolution, hindering its application and development. For these reasons, it is of great significance to fill this gap in genetic information to facilitate research on P. hirtellum.
The chloroplast (cp) genome provides molecular markers that are an excellent tool for phylogenetic analyses from specifications to taxa at higher ranks (Zoschke and Bock, 2018; Qu et al., 2023), which has been successfully used to elucidate genetic and phylogenetic information of certain species, such as Camellia, Aruncus, and Neocinnamomum (Lin et al., 2022; Park et al., 2022; Cao et al., 2023). However, it is essential to note that the cp gene approach may have limitations, such as missing relationships due to length variations, gap/index deletions, and incorrect models of sequence evolution in condensed datasets (Goremykin et al., 2005). On the other hand, the coding sequence (CDS) is relatively stable and can collaborate with cp genomes to construct plant phylogenetic relationships for more accurate results (Chen et al., 2023; Jiang et al., 2022). Herein, the cp genome of P. hirtellum was sequenced, the characters were elucidated, and a phylogenetic tree was constructed based on the cp genome and CDS. This study thus provides a scientific foundation for the conservation of P. hirtellum.
MATERIALS AND METHODS
Fresh and healthy leaves of P. hirtellum were collected from Yanjing County in Tibet, China. The voucher specimen (YN2022HJ455) were identified by Prof. Baozhong Duan and were preserved at the herbarium of Dali University. An amount of approximately 1.0 g of fresh leaves was collected, immediately frozen in liquid nitrogen, and stored for subsequent DNA extraction. Genomic DNA was extracted using the Plant Genomic DNA kit (Tiangen, Beijing, China) following the manufacturer's instructions. The quality and quantity of the extracted DNA were evaluated using a high-sensitivity Qubit 4.0 Fluorometer (Life Technologies, Inc., Carlsbad, CA, USA).
To prepare the sequencing libraries, a high-quality DNA sample of at least 30 μL was utilized, with a minimum concentration of 100 ng/μL. The libraries were then sequenced using the Illumina NovaSeq system (Illumina, San Diego, CA, USA). The paired-end sequence reads were filtered to trim low-quality bases and adapter sequences using Toolkit v2.3.3 software. The clean data was assembled using GetOrganelle v.1.6.4, exploiting Bowtie2 v.2.4.4, SPAdes v.3.13.0, and Blast v.2.5.0 as dependencies (Jin et al., 2019). Following the assembly step, two online annotation tools, CpGAVAS2 (http://47.96.249.172:16019/analyzer/annotate) (An et al., 2020) and GeSeq (https://chlorobox.mpimp-golm.mpg.de/geseq.html) (Castro et al., 2023), were employed to annotate the circular cp genomes. The annotated cp genome sequence was deposited into the GenBank database of the National Center for Biotechnology Information (NCBI), with accession number OR492287. Gene maps were visualized using the OGDRAW tool (https://chlorobox.mpimp-golm.mpg.de/OGDraw.html). Geneious 9.0.2 software was used to analyze the GC content, genome size, tRNA, and repeat content. Moreover, CodonW v.1.4.2 was employed to determine the amino acid usage frequency and relative synonymous codon usage (RSCU) (Sharp et al., 1986).
Four types of dispersed repeat sequences, Forward (F), Reverse (R), Palindromic (P), and Complementary (C), were identified using the REPuter tool (https://bibiserv.cebitec.unibielefeld.de/reputer/) with a minimum repeat size of 20 bp and a similarity threshold of 90% between repeat pairs (Zhou et al., 2022). In addition, simple sequence repeats (SSRs) were analyzed using MISA software (http://pgrc.ipk-gatersleben.de/misa/) (Beier et al., 2017), with thresholds of ‘10’ for mono, ‘5’ for di-, ‘4’ for tri-, and ‘3’ for tetra-, penta-, and hexanucleotide motifs. The IRSCOPE (https://irscope.shinyapps.io/irapp/) online tool was used to analyze boundary information from the cp genome of P. hirtellum.
A total of 38 taxa, comprising one cp genome annotated by our study and 37 cp genomes obtained from the NCBI, were selected to reconstruct the phylogenetic relationships. Two species, Heteropolygonatum ogisui (GenBank MZ150833) and H. alternicirrhosum (GenBank MZ150832) were chosen as outgroups. Additionally, CDS were extracted from 38 cp genomes. The cp genomes and CDS were aligned using the MAFFT program and were verified manually. The maximum likelihood (ML) tree was reconstructed using IQtree with default settings, i.e., 1,000 iterations, 1,000 replications, and the best-fit model selection (Katoh and Standley, 2013).
RESULTS AND DISCUSSION
The raw data of P. hirtellum were filtered to remove adaptors and low-quality reads. After assembly and splicing, the cp genomes were obtained. The result showed that the cp genome of P. hirtellum was a small circular DNA molecule and exhibited a typical quadripartite structure consisting of large single copy (LSC), small single copy (SSC), and two inverted repeat (IR) regions, as shown in Fig. 1. The total length of the cp genome is 155,697 bp, including 84,468 bp for LSC, 18,491 bp for SSC, and 26,369 bp for IR. In addition, the total GC content of the cp genome amounted to 37.7%, and there were significant differences in the GC contents in the LSC, SSC, and IR regions. As shown in Table 1, the GC content of the IR region was highest (43.0%), followed by LSC (35.7%) and SSC (31.6%), likely because the IR region contains rRNA genes with a high GC content (Wu et al., 2020).

Chloroplast genome annotation of Polygonatum hirtellum as drawn by OGDRAW.

Base composition of the cp genome in Polygonatum hirtellum.
One-hundred and thirty genes were identified from the cp genomes of P. hirtellum, consisting of 84 protein-coding genes, 38 tRNA genes, and eight rRNA genes. These genes could be categorized into the following four major groups: photosynthesis-related genes, replication-related genes, protein genes, and other genes (Table 2), of which 19 were repeated as members of IR regions, consisting of six protein-coding genes (rpl2, rpl23, rps7, rps19, ndhB, and ycf2), eight tRNA genes (trnH-GUG, trnI-CAU, trnL-CAA, trnV-GAC, trnI-GAU, trnA-UGC, trnR-ACG, and trnN-GUU), four rRNA genes (rrn16S, rrn23S, rrn4.5S, and rrn5S), and a trans-spliced gene, rps12. Additionally, seven genes in P. hirtellum contained introns, of which two genes (ycf3 and petB) contained two introns and the remaining five only contained a single intron.
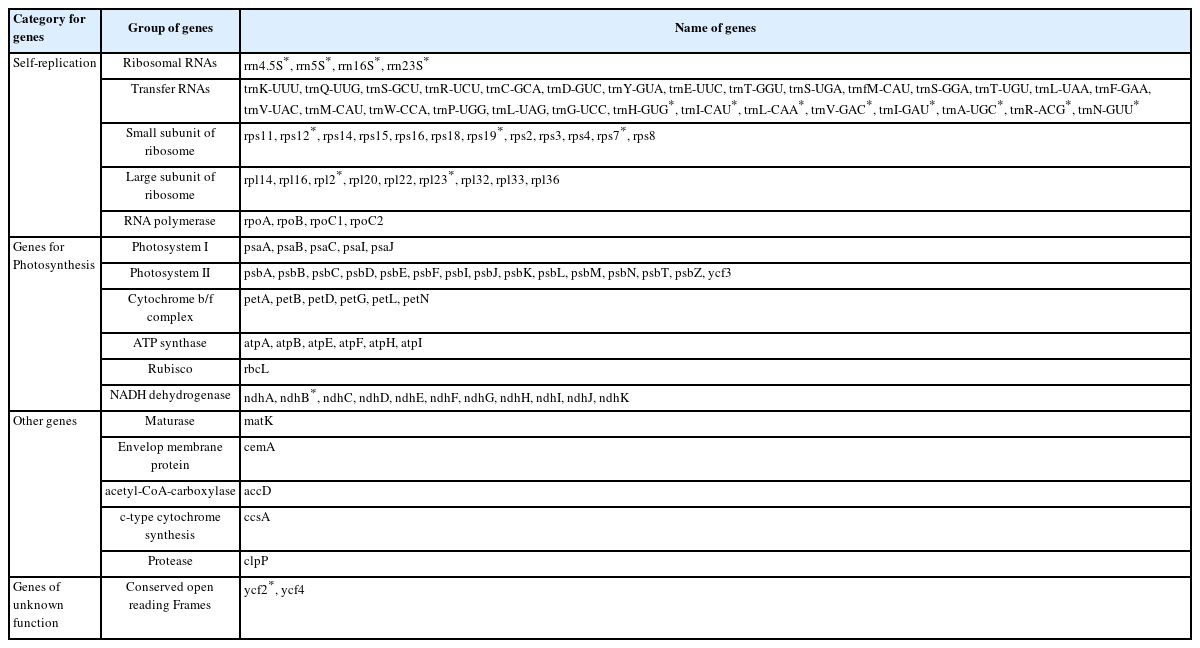
Gene composition in the Polygonatum hirtellum cp genome.

Number of types of repeat sequences found in the Polygonatum hirtellum cp genome, where R, P, F, and C indicate the repeat types.
The cp genome of P. hirtellum was analyzed to investigate the amino acid frequency, number, the codon usage bias, and RSCU. The results showed that the cp genome sequences encoded 21 amino acids, and 64 codons were deduced.
Among the amino acids encoded by the cp genome's genes, leucine (Leu) was most frequently occurring, encoding 2015 times and contributing 10.31% of the total. Conversely, cysteine (Cys) emerged as a relatively infrequent amino acid, coding a mere 220 times and accounting for 1.13%. Similar results have been documented within other members of the Polygonatum genus (Liu et al., 2022; Shi et al., 2022). Moreover, pronounced bias toward A/T was observed at the third position of the codon. Similar results were observed in other angiosperm taxa (Jiang et al., 2022; Meng et al., 2023).
As shown in Fig. 2, the RSCU values of the 64 codons were identified. The results demonstrated that AUU exhibited the highest frequency, followed by GAA, while UGC was the least prevalent. Notably, the unique codon UGG for tryptophan, AUG for methionine, and UCC for serine were excluded from the analysis due to their lack of a clear discernible trend, as indicated by an RSCU value of 1. Moreover, 30 codons were identified in the high-frequency cohort (RSCU > 1), of which 29 codons ended in A/U, with the exception of UUG. Concurrently, within the low-frequency assemblage (RSCU < 1), consisting of 31 codons, 28 codons were observed to conclude in nucleotide residues C/G, with the exceptions being UGA, CUA, and AUA.
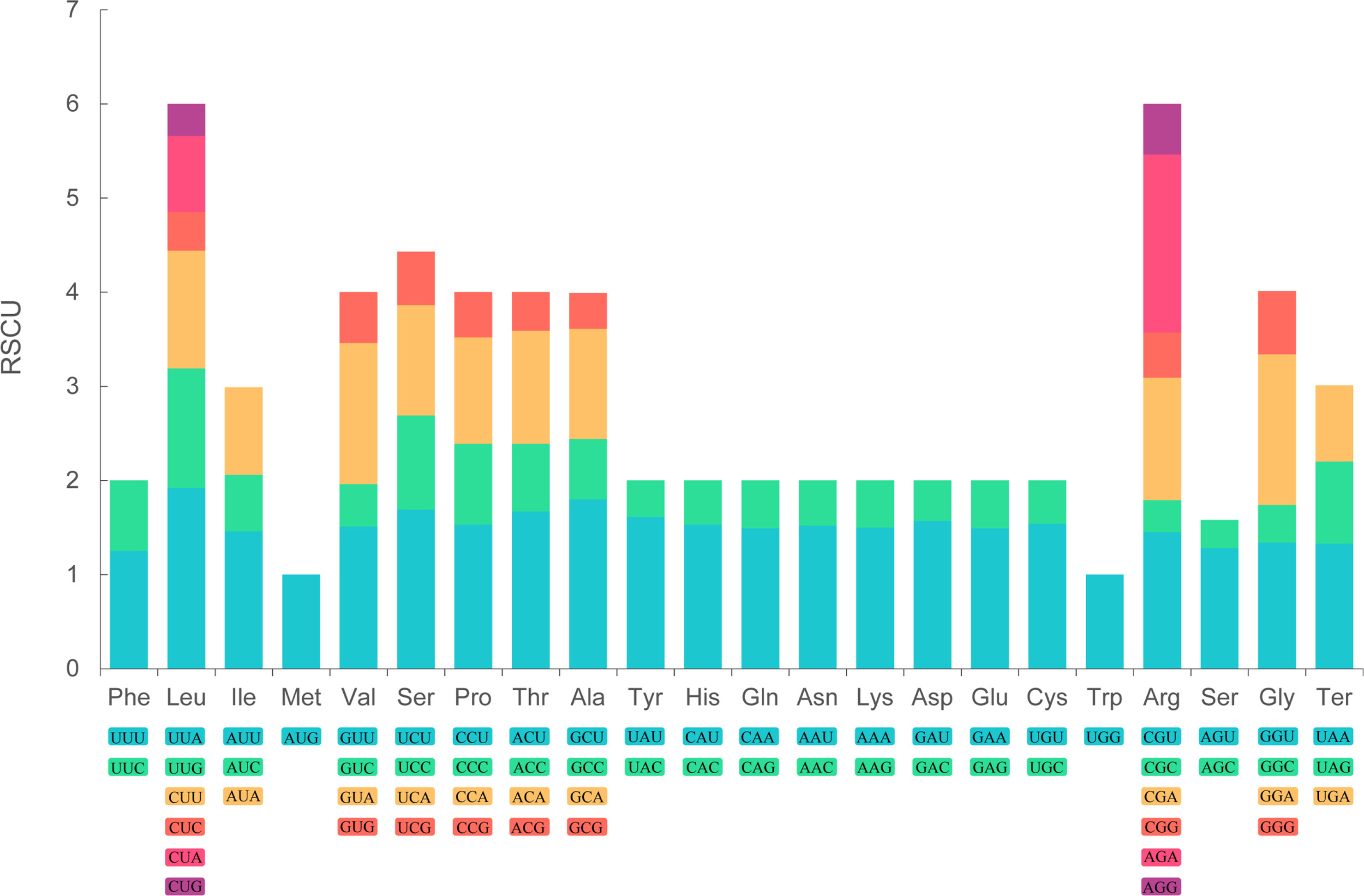
Codon content of 21 amino acids and stop codons in all protein-coding genes of the chloroplast genome of Polygonatum hirtellum. RSCU, relative synonymous codon usage.
Additionally, 99 dispersed repeat sequences, consisting of two C, 12 R, 36 F, and 49 P, were identified in the cp genome of P. hirtellum, and the lengths of the repeat sequences ranged from 20 to 71 bp, with 20–39 bp being the most frequent and those longer than 50 bp being the least abundant. The F and P repeat sequences were more abundant than the R and C repeats.
SSRs are tandem repeats of 1–6 nucleotide motifs, which can be used for species identification and genetic diversity research due to their rich polymorphism, site specificity, multiple alleles, and reliability (An et al., 2020; Idrees and Irashad, 2014). Here, 55 SSRs were identified in the cp genome of P. hirtellum, which could be classified into mono-nucleotide repeat sequences (A/T), di-nucleotide repeat sequences (AG/CT and AT/CG), tri-nucleotide repeat sequences (AAT/ATT and AGC/CTG), and tetra-nucleotide repeat sequences (AAAT/ATTT, AATC/ATTG, and AATG/ATTC) based on the number of nucleotides. Mono-nucleotide repeats were the most abundant among these SSRs, followed by di-nucleotide repeats. In addition, A and T bases were found to be the basic repeat units within most SSRs, and this result implied a high A/T preference in the cp genome of P. hirtellum, in good agreement with observations of angiosperm cp genomes (Guan et al., 2022).
The analysis also showed that SSRs were mainly located in the LSC regions rather than in the IR and SSC regions. Notably, most SSRs were located in the coding region, followed by the intergenic spacer, exons, and introns region. The number of SSRs was lower than in the non-coding region, differing from the previously reported cp genome characteristics of other species in the genus Polygonatum (Wang et al., 2022). This discrepancy is attributable to the significant selective pressure experienced by the non-coding regions of P. hirtellum, resulting in higher genetic diversity relative to that of the coding regions (Kelchner, 2000; Shaw et al., 2014).
The cp genome of P. hirtellum exhibited four boundaries: LSC-IRb, IRb-SSC, SSC-IRa, and IRa-LSC (Fig. 3). The rps19 gene was located in the IRa region, 37 bp from the IRa/LSC border. Similarly, the rpl22 gene was entirely situated in the LSC region, with a distance of 8 bp from the LSC/IRb border. The ndhF gene was found at the junction of IRb/SSC, extending 31 bp into the IRb region.

Inverted repeats of Polygonatum hirtellum.
Cp genomes are a valuable source of phylogenetic information and are commonly used for reconstructing phylogenies and analyzing plant populations (Su et al., 2023; Yan et al., 2019). In this study, we constructed phylogenetic trees using both the cp genomes and CDS of 35 species of Polygonatum from the NCBI database. As illustrated in Fig. 4, ML analyses showed that P. hirtellum was a member of the section Verticillata and formed an evolutionary branch with multiple species, such as P. stewartianum, P. kingianum, and P. stenophyllum. Bootstrap value (82) suggested a significant difference between P. hirtellum and other species of the genus Polygonatum used in this analysis. This result is further supported by the phylogenetic trees constructed based on CDS, as shown in Fig. 5.
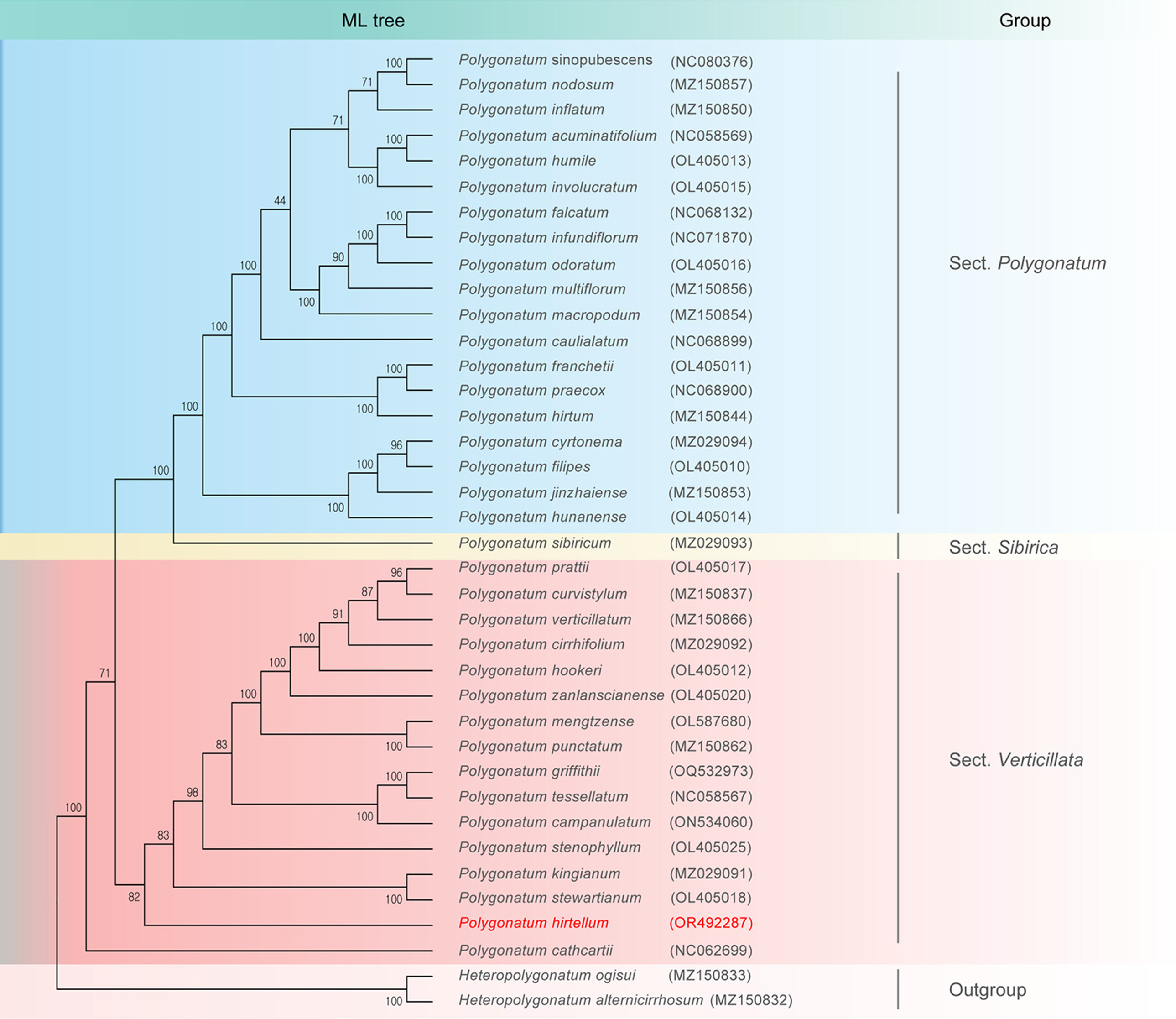
Phylogenetic tree based on chloroplast genome sequences according to a maximum likelihood (ML) method. Bootstrap support values are presented on each branch.

Phylogenetic tree based on coding sequences according to maximum likelihood (ML) method. Bootstrap support values are presented on each branch.
In summary, this investigation reveals the cp genome and phylogenetic relationships of P. hirtellum, offering valuable insights into its genetic diversity and making significant contributions to its conservation.
Acknowledgements
This work was supported by the Yunnan academician expert workstation (202105AF150053), key technology projects in Yunnan province of China (202002AA100007), and by the Yunnan Xingdian talent support plan (YNWR-QNBJ-2020251). We thank Northeast Forestry University and the China Academy of Chinese Medical Sciences for their technical assistance.
Notes
CONFLICTS OF INTEREST
The authors declare that there are no conflicts of interest.