The complete chloroplast genome of Campsis grandiflora (Bignoniaceae)
Article information

Abstract
Campsis grandiflora (Thunb.) K. Schum is an ornamental species with various useful biological effects. The chloroplast genome of C. grandiflora isolated in Korea is 154,293 bp long (GC ratio: 38.1%) and has four subregions: 84,121 bp of large single-copy (36.2%) and 18,521 bp of small single-copy (30.0%) regions are separated by 24,332 bp of inverted repeat (42.9%) regions including 132 genes (87 protein-coding genes, eight rRNAs, and 37 tRNAs). One single-nucleotide polymorphism and five insertion and deletion (INDEL) regions (40-bp in total) were identified, indicating a low level of intraspecific variation in the chloroplast genome. All five INDEL regions were linked to the repetitive sequences. Seventy-two normal simple sequence repeats (SSRs) and 47 extended SSRs were identified to develop molecular markers. The phylogenetic trees of 29 representative Bignoniaceae chloroplast genomes indicate that the tribe-level phylogenic relationship is congruent with the findings of previous studies.
INTRODUCTION
Genus Campsis Lour. consists of only two species, Campsis grandiflora (Thunb.) distributed in East Asia and Campsis radicans (L.) Bureau found in North America (Wen and Jansen, 1995). Because of its disjunct distribution, this genus was considered as a material to understand their evolutionary history, resulting that both species was estimated to be diversified at 24.4 million years ago (Wen and Jansen, 1995). Campsis grandiflora has been utilized as an ornamental species because of their trumpet shape flowers (Jia et al., 2012). Moreover, C. grandiflora was known to have various biological effects (Yu et al., 2015; Oku et al., 2019), such as anti-oxidative and anti-inflammatory (Cui et al., 2006), and useful phytocompounds (Jin et al., 2005; Kim et al., 2007; Han et al., 2012) including triterpenoids (Kim et al., 2005). To understand intraspecific variations of C. grandiflora chloroplast genome together with the previously published chloroplast genome isolated in China (Chen et al., 2022), we completed the chloroplast genome of C. grandiflora isolated in Korea.
MATERIALS AND METHODS
Plant material
The sample was collected on Gangseo postal office, Seoul, Korea (37.565175N, 126.840624E). A specimen was deposited at the InfoBoss Cyber Herbarium (IN) under the voucher number, IB-01065. No permission is required for the collection.
DNA extraction and chloroplast genome determination
Total DNA was extracted from the fresh leaves using a DNeasy Plant Mini Kit (Qiagen, Hilden, Germany). The sequencing library was constructed using an Illumina TruSeq Nano DNA Library Preparation Kit (Illumina, San Diego, CA, USA) following the manufacturer’s recommendations with approximately 350-bp DNA fragments. 4.15-Gbp raw sequences were obtained using NovaSeq6000 at Macrogen Inc., Korea, and were filtered by Trimmomatic v0.33 (Bolger et al., 2014). The chloroplast genome was de novo assembled with Velvet v1.2.10 (Zerbino and Birney, 2008), and gaps were closed using GapCloser v1.12 (Zhao et al., 2011). The genome sequence was confirmed by aligning all raw reads against the assembled genome using BWA v0.7.17 and SAMtools v1.9 (Li et al., 2009; Li, 2013). All processes were conducted in the Genome Information System (http://geis.infoboss.co.kr) utilized in previous studies (Kim et al., 2019e, 2020; Park et al., 2019c; Park and Xi, 2021). Geneious Prime v2020.2.4 (Biomatters Ltd., Auckland, New Zealand) was used for annotation based on the Tecomaria capensis chloroplast genome (GenBank accession number: NC_037462) (Fonseca and Lohmann, 2018). A circular map of C. grandiflora chloroplast genome was drawn using OGDRAW v1.31 (Greiner et al., 2019). Large single-copy (LSC), small single-copy (SSC), and inverted repeat (IR) regions were determined by bl2seq (Tatusova and Madden, 1999).
Identification of intraspecific variations
Single nucleotide polymorphisms (SNPs) and insertions and deletions (INDELs) were identified from the pair-wise sequence alignment of the two C. grandiflora chloroplast genomes conducted by MAFFT 7.450 (Katoh and Standley, 2013) with ‘Find variations/SNPs’ implemented in Geneious Prime 2020.2.4 (Biomatters Ltd.), which has been used in the previous studies investigating intraspecific variations on organelle genomes (Kim et al., 2021a; Oh et al., 2021; Suh et al., 2021). INDEL region was defined as the continuous INDELs.
Identification and comparative analysis of simple sequence repeats
Simple sequence repeats (SSRs) were identified on the chloroplast genome sequence using the pipeline of the SSR database (SSRDB; http://ssrdb.infoboss.co.kr/) which has been utilized in several organelle genomic studies (Lee et al., 2020; Choi et al., 2021; Park et al., 2021d, 2022). The SSR is conventionally recognized as the nucleotide array composed of repeats with one or up to six base pair units. For example, monoSSR refers an array of nucleotide repeats containing a particular base and hexaSSR an array of nucleotide repeats containing six base pair unit. The overall length of SSR is mostly over 10 base pairs. In this study, we tried to classify SSR with more criteria which has been applied in previous analyses (Gandhi et al., 2010; Chen et al., 2015; Cheng et al., 2016; Shukla et al., 2018; Jeon and Kim, 2019; Li et al., 2019). The criteria applied are (1) ‘normal SSR’ as a conventional definition from monoSSR to hexaSSR, (2) ‘extented SSR’ referring from heptaSSR (repeats of 7 bp unit) to decaSSR (repeats of 10 bp unit), and (3) ‘potential SSR’ referring specific cases with only 2 units in pentaSSR and hexaSSR. These criteria have been applied and provided better understanding of SSR patterns in previous analyses in chloroplast genomes of Dysphania species (Kim et al., 2019f), Arabidopsis thaliana (Park et al., 2020c), Chenopodium album (Park et al., 2021b), Diarthron linifolium (Kim et al., 2021b), and mitochondrial genome of Rosa rugosa (Park et al., 2020d).
Phylogenetic analysis
Twenty-nine representative Bignoniaceae chloroplast genomes including two C. grandiflora chloroplast genomes and one outgroup species, Paulownia tomentosa (Yi and Kim, 2016), were used for calculating multiple sequence alignments of 60 conserved genes by MAFFT v7.450 (Katoh and Standley, 2013) for constructing phylogenetic trees. We used MEGA X (Kumar et al., 2018) to construct maximum likelihood (ML) and neighbor-joining (NJ) and MrBayes v3.2.6 (Ronquist et al., 2012) to carry out Bayesian inference (BI). A heuristic search was used with nearest-neighbor interchange branch swapping, the Tamura-Nei model, and uniform rates among sites to construct ML and NJ phylogenetic trees with default values for other options. To estimate the node confidences bootstrap analyses with 1,000 and 10,000 bootstrap pseudoreplicates were conducted for ML and NJ trees, respectively. For the BI analysis, the GTR (general time reversible) model with gamma rates was used as a molecular model and Markov-Chain Monte Carlo algorithm was employed for 1,000,000 generations with four chains running simultaneously. To build the consensus tree of BI, we sampled trees every 200 generations after removing 100,000 generations as a ‘burn-in’.
RESULTS AND DISCUSSION
The chloroplast genome of C. grandiflora (GenBank accession number: OM279807) is 154,293 bp (GC ratio: 38.1%) and has four subregions: 85,078 bp of LSC region (36.2%) and 18,577 bp of SSC region (33.0%) regions are separated by 25,319 bp of IR region (43.2%) (Fig. 1). Its length is shorter than that of the previous chloroplast genome by 10 bp (154,303 bp; GenBank accession number: MW430049). It contains 132 genes (87 protein-coding genes [PCGs], eight ribosomal RNAs [rRNAs], and 37 transfer RNAs [tRNA]); 19 genes (eight PCGs, four rRNAs, and seven tRNAs) are duplicated in IR regions (Fig. 1). Structural variation between C. grandiflora and T. capensis was identified using Mauve v1.1.3 (Darling et al., 2004) in LSC region: the region between 48,536 bp and 73,124 bp in C. grandiflora chloroplast genome was inverted against that of T. capensis. This phenomenon also occurred between two Incarvillea chloroplast genomes in the same family (Ma et al., 2019; Wu et al., 2021), congruent to the previous study (Chen et al., 2022). It suggests that inversion events in LSC occurred in Bignoniaceae in comparison to the other families, such as Amaranthaceae (Park et al., 2021b) and Oleaceae (Park et al., 2019f).
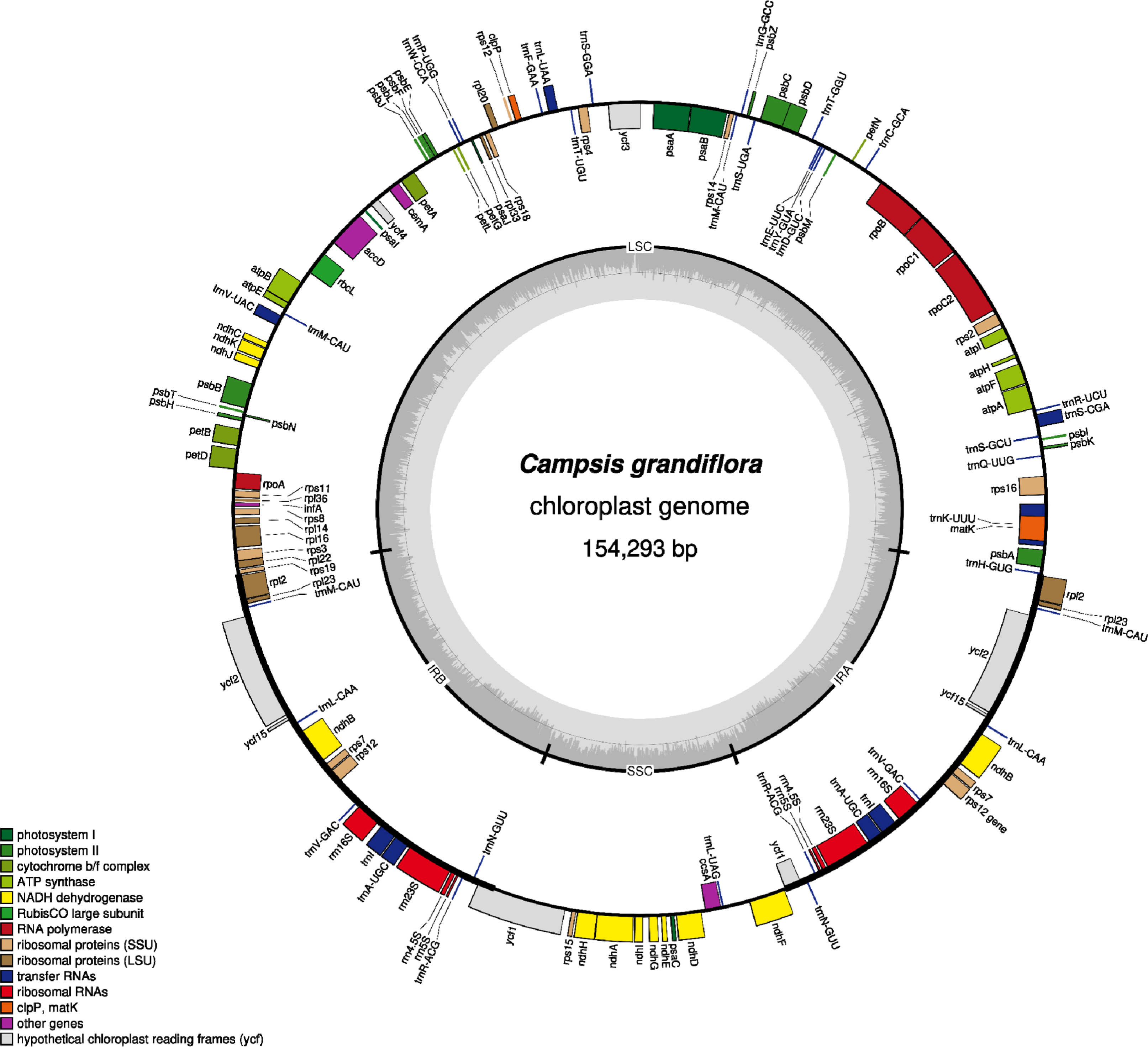
Circular map of Campsis grandiflora complete chloroplast genome. The genes located outside of the circle are transcribed clockwise, while those located inside are transcribed counterclockwise. The dark gray plot in the inner circle corresponds to GC content. Large singlecopy (LSC), small single-copy (SSC), and inverted repeat (IR) are indicated with LSC, SSC, and IR (IRA and IRB), respectively.
Interspecific variations between the two C. grandiflora chloroplast genomes were investigated. In total, one SNP and five INDEL regions (40 bp in total). One SNP was located between trnC and petN. The 20-bp deletion, which is the longest INDEL, was found in 3' end of ycf1, which expanded two more amino acids (Fig. 2A). Another 15-bp INDEL region was located in the first intron of accD, presenting the three-time repeat in the chloroplast genome assembled in this study while two-time repeat in the previous chloroplast genome (Fig. 2B). One-bp deletion was found in the intergenic region between trnK and rps16, exhibiting difference of monoSSR (Fig. 2C). The two INDEL regions were found in 16S rRNA in the IR region (Fig. 2D), showing that two-time repeat of CAT was destroyed in the chloroplast genome assembled in this study by this 2-bp deletion (Fig. 2D). Interestingly, all INDEL regions were linked to the repetitive sequences and proportion of INDEL regions related to SSRs (20%) was low. Numbers of intraspecific variations of C. grandiflora are relatively lower than those identified between the samples between Korea and China (Fig. 2E, Table 1). This result seems to be incongruent to the previous studies that estimated their genetic diversities using the classical methods (He and Gu, 1990; Wu et al., 1990; Wen and Jansen, 1995). Therefore, additional C. grandiflora chloroplast genomes will be required to evaluate its genetic diversity.
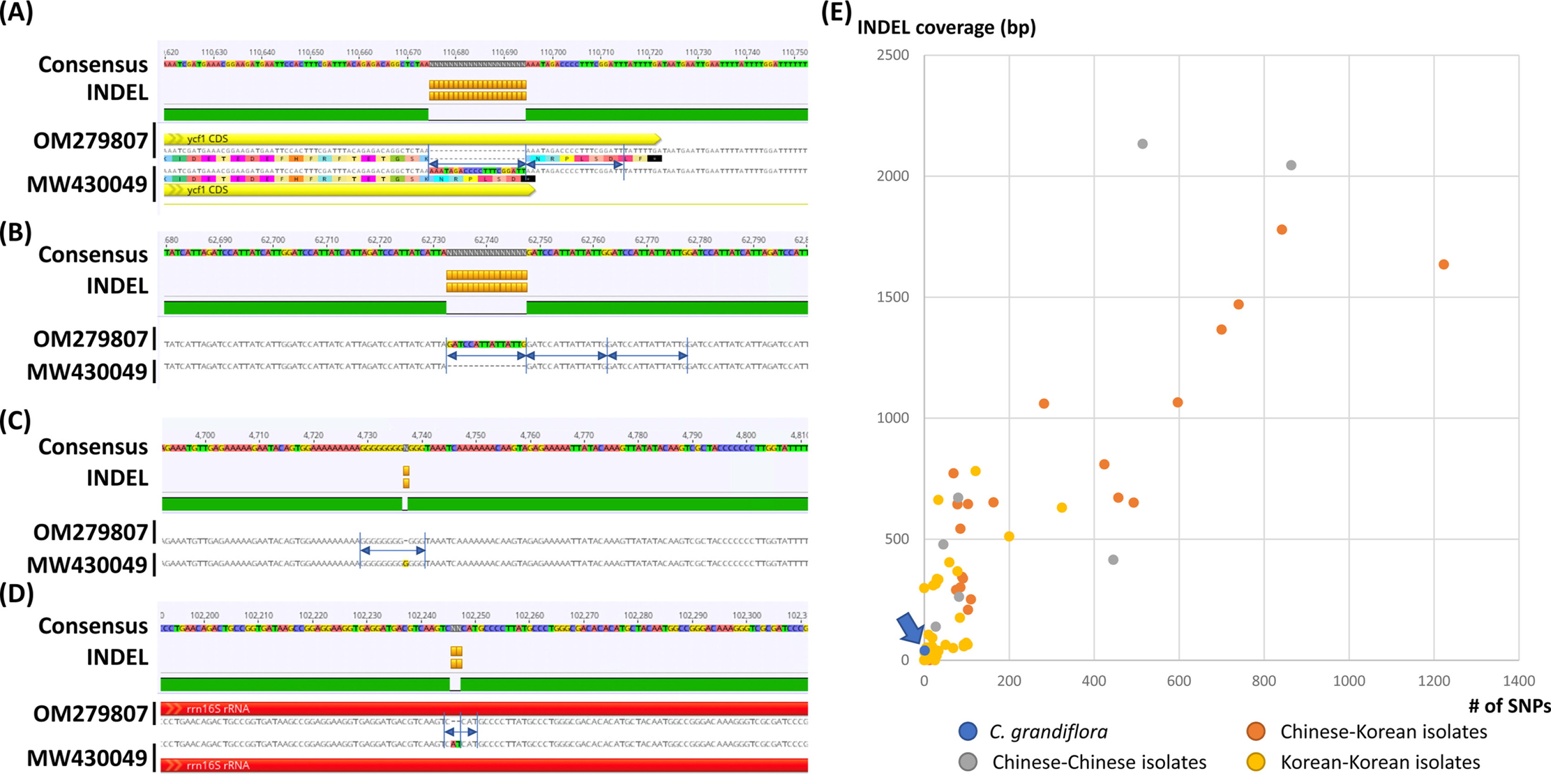
Intraspecific variations identified from the two Campsis grandiflora chloroplast genomes. A–D. Consensus sequences were displayed with different background colors of each base. Insertions and deletions were presented as orange-colored boxes. Yellow and red arrows indicate protein-coding genes (PCGs) and rRNAs, respectively. Amino acid sequences in PCG were displayed below the nucleotide sequences. Blue arrows and lines indicate repetitive sequences found in insertions and deletions. E. X-axis means the number of single nucleotide polymorphism (SNPs) and Y-axis indicates insertion and deletion (INDEL) coverage (bp). Blue-colored circle indicates the number of intraspecific variations identified in C. grandiflora chloroplast genomes. Orange, gray, and yellow-colored circles presented those identified between Chinese and Korean isolates, Chinese and Chinese isolates, and Korean and Korean isolates, respectively.

List of numbers of intraspecific variations in the chloroplast genomes identified from the samples isolated from Korea and China.
SSR has been utilized as useful molecular markers (Huang et al., 2015; Li et al., 2020a, 2020b). Seventy-two normal SSRs, 418 potential SSRs, and 47 extended SSRs were identified in both C. grandiflora chloroplast genomes (Table 2). Most of normal SSRs are monoSSRs (Fig. 3), which is similar to those of the other plant species (Kim et al., 2019f, 2021b; Park et al., 2020c, 2021b). Nine normal SSRs and 18 extended SSRs (22.68%) were identified in the genic regions of matK, atpA, rpoC2, psbC, psaI, psbB, rpoA, rpl22, ycf2, ycf1, ndhI, ndhG, and ndhD (Table 2) and 12 normal SSRs and three extended SSRs (12.61%) were found in the intronic regions of rps16, trnS-CGA, atpF, ycf3, trnL-UAA, petD, rps16, and ndhA (Table 2). Due to low number of intraspecific variations, only one monoSSR (cM0000002) displayed the differences of the number of repeats between the two chloroplast genomes. These SSRs will be useful to develop molecular makers because high genetic diversity of C. grandiflora was estimated in previous studies (He and Gu, 1990; Wu et al., 1990; Wen and Jansen, 1995).

List of normal SSRs and extended SSRs identified in the C. grandiflora chloroplast genomes isolated in Korea.
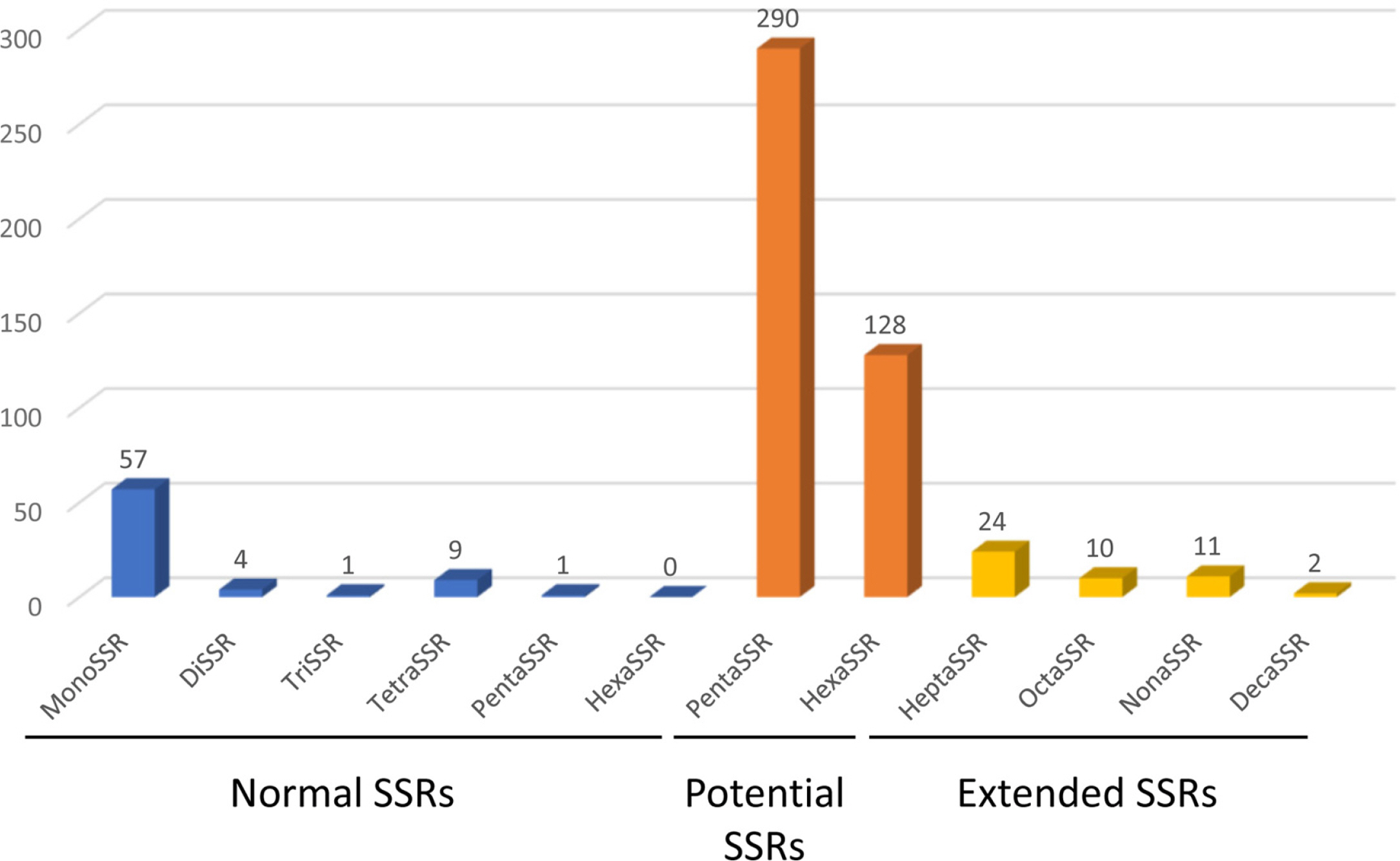
Distribution of simple sequence repeats (SSRs) along with types identified from Campsis grandiflora chloroplast genome. X-axis presented SSR types of normal SSRs, potential SSRs, and extended SSRs. Y-axis indicates the number of SSRs.
Three phylogenetic trees showed that C. grandiflora was clustered with T. capensis/ Incarvillea with high supportive values (Fig. 4). In addition, trees presented that tribes covering more than one chloroplast genome, including Tecomeae, Catalpeae, Crescentiina, and Bignonieae, were well clustered with high supportive values (Fig. 4). It is congruent to previous phylogenetic studies, except for the Catalpeae and Oroxylae clustered in one clade with week bootstrap values (Olmstead et al., 2009). It may be caused by different coverage of samples between the two studies. Together with additional chloroplast genomes of Bignoniaceae, C. grandiflora chloroplast genome will help to understand evolutionary history of Bignoniacea.
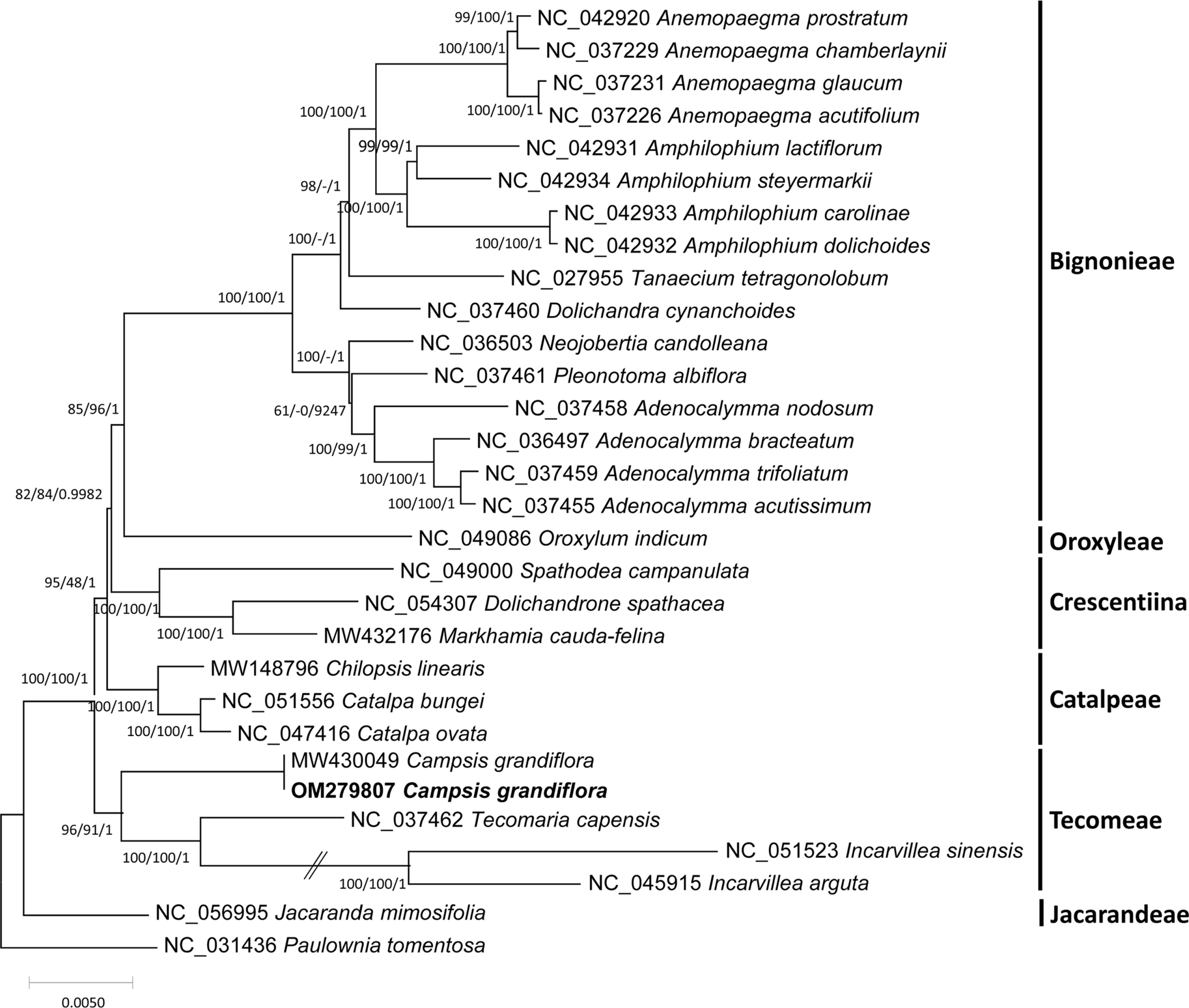
Phylogenetic trees of the 29 representative Bignoniaceae chloroplast genomes including two Campsis grandiflora chloroplast genomes. Phylogenetic tree was drawn based on the maximum likelihood phylogenetic tree. The numbers above the branches correspond to the bootstrap support values from the maximum likelihood and neighbor-joining methods, as well as posterior probability from the Bayesian inference.
Acknowledgements
This study was supported by the InfoBoss Research Grant (IBG-0038).
Notes
CONFLICTS OF INTEREST
The authors declare that there are no conflicts of interest.