The complete chloroplast genome of Diarthron linifolium (Thymelaeaceae), a species found on a limestone outcrop in eastern Asia
Article information

Abstract
Diarthron linifolium Turcz. is an annual herb usually found in sandy soil or limestone areas. Plants in the genus Diarthron are known to have toxic chemicals that may, however, be potentially useful as an anticancer treatment. Diarthron linifolium is a unique species among the species of the genus distributed in Korea. Here, we determine the genetic variation of D. linifolium collected in Korea with a full chloroplast genome and investigate its evolutionary status by means of a phylogenetic analysis. The chloroplast genome of Korean D. linifolium has a total length of 172,644 bp with four subregions; 86,158 bp of large single copy and 2,858 bp of small single copy (SSC) regions are separated by 41,814 bp of inverted repeat (IR) regions. We found that the SSC region of D. linifolium is considerably short but that IRs are relatively long in comparison with other chloroplast genomes. Various simple sequence repeats were identified, and our nucleotide diversity analysis suggested potential marker regions near ndhF. The phylogenetic analysis indicated that D. linifolium from Korea is a sister to the group of Daphne species.
Diarthron linifolium Turcz. is an annual herb mainly distributed in eastern Asia including western China, Mongolia, Far East Russia, and Korea. Plants belonging to D. linifolium are usually found in sandy soil or limestone area. Plants of Diarthron are known to be very toxic (Tan, 1982). It has been shown that species of Diarthron contain sesquiterpenoids in the roots with antineoplastic activities, suggesting that they may be useful for anticancer treatment (Sun et al., 2018). The Korean plants of D. linifolium are the southernmost population of the species and isolated from its main distribution range. It is recently designated as a vulnerable species listed by the Korean government (https://species.nibr.go.kr) because it is hardly found, and its distribution is confined in limestone area. Thus, detailed understanding of genetic diversity and biogeographic origin is needed for the conservation of the species.
Tan (1982) summarized the taxonomic status of the genus Diarthron by suggesting new combinations of taxonomic ranks and subgroups representing three subgenera, Diarthron (two species in two sections, Diarthron and Arthrochlamys), Dendrostellera (eight species), and Stelleropsis (nine species in two sections, Stelleropsis and Turcomanica), which are mainly distinguished from habit (herbaceous vs. suffrutescent or shrubby) or floral morphology (sericeous-villous vs. glabrous or sparsely pubescent of perianth) (Tan, 1982). Diarthron linifolium is similar to Diarthron vesiculosum (Fisch. & C. A. Mey. ex Kar. & Kir.) C. A. Mey. in gross morphology and habit in that both species are annual herb with slender stems and racemes but distinctly recognized by the number of stamens, 4 or 5 in D. linifolium but 8 in D. vesiculosum. Interestingly, the distribution of D. linifolium is not overlapped that of D. vesiculosum that is more concentrated in northwestern Central Asia.
The phylogenetic position of Diarthron including D. linifolium is not fully assessed so far. The close relationship of Diarthron to Stellera was proposed based on the gross morphology (Tan, 1982) and molecular phylogeny (Galicia-Herbada, 2006) but other molecular phylogenetic studies with species in the family, Thymelaeaceae, suggested that Diarthron is more closely related to Daphne and Thymelaea with its sister relationship to Stellera and Wikstroemia (Van der Bank et al., 2002; Beaumont et al., 2009). In contrast, Qian et al. (2021) suggested closer relationship of Diarthron to Stellera and Wikstroemia with moderate bootstrap support and posterior probability from nuclear rDNA internal transcribed spacer (ITS) sequence analysis (Qian et al., 2021).
Recent rapid development of next-generation and third-generation sequencing technologies (Roberts et al., 2013; Deamer et al., 2016; Goodwin et al., 2016) have facilitated organelle genome research, resulting in more than 10,000 chloroplast genomes in National Center for Biotechnology Information (NCBI). It indicates that chloroplast genomes of closely related species within the family will be enough to conduct comparative analysis of Diathron. At least 29 chloroplast genomes of Thymelaeaceae are available (Supplementary Table 1), which is favorable to conduct comparative analysis using chloroplast genome of D. linifolium. As part of genetic and biogeographic study of D. linifolium, we first determined and presented the complete chloroplast genome of D. linifolium in this study and conducted comparative analyses.
Materials and Methods
De novo assembly and annotation of Diarthron linifolium chloroplast genome
The sample of D. linifolium for DNA isolation was collected at the limestone beds in the lakeshore of the Lake Cheongpungho, Jecheon-si, Chungcheongbuk-do, Korea (37º1′44″N, 128º11′15″E). Voucher specimen was deposited in the herbarium of Daejeon University (TUT) under the number Oh 8252. Total DNA was extracted from fresh leaves using DNeasy Plant Mini Kit (QIAGEN, Hilden, Germany). The DNA library for the high-throughput short read sequencing was prepared with the protocol provided by the Illumina TruSeq DNA Library Prep Kit v2 (Illumina, San Diego, CA, USA) and the whole genome sequencing was conducted with Illumina NovaSeq6000 at Macrogen, Korea. 6.9-Gbp raw reads were used for chloroplast de novo genome assembly with Velvet v1.2.10 (Zerbino and Birney, 2008) after filtering raw reads using Trimmomatic v0.33 (Bolger et al., 2014). After getting first draft of chloroplast genome sequences, gap was filled with GapCloser v1.12 (Zhao et al., 2011) and all bases from the assembled sequences were confirmed using BWA v0.7.17 (Li, 2013) and SAMtools v1.9 (Li et al., 2009). All bioinformatic processes for de novo assembly was conducted by the pipeline, Genome Information System (GeIS; http://geis.infoboss.co.kr), which has been efficiently utilized and optimized in the previous studies (Min et al., 2019; Yun et al., 2019; Kim et al., 2021a, 2021b; Park et al., 2021b).
Geneious Prime v2020.2.4 software (Biomatters Ltd, Auckland, New Zealand) was used for chloroplast genome annotation for which we utilized information from Daphne genkwa chloroplast genome (MT754180) (Yoo et al., 2021) by transferring annotations with corrections of exceptional cases including missing start or stop codons. tRNA was predicted and confirmed using tRNAScan-SE v2.0.6 (Schattner et al., 2005).
Identification of simple sequence repeats on D. linifolium chloroplast genome
Simple sequence repeats (SSRs) were identified on the chloroplast genome sequence using the pipeline of the SSR database (SSRDB; http://ssrdb.infoboss.co.kr/) which has been utilized in various organelle genomic researches (Kim et al., 2019; Lee et al., 2020; Park et al., 2020a, 2021a; Choi et al., 2021). The SSR is conventionally recognized as the nucleotide array composed of repeats with one or up to six base pair units. For example, monoSSR refers an array of nucleotide repeats containing a particular base and hexaSSR an array of nucleotide repeats containing six base pair unit. The overall length of SSR is mostly over 10 base pairs. In this study, we tried to classify SSR with more criteria which has been applied in previous analyses (Gandhi et al., 2010; Chen et al., 2015; Cheng et al., 2016; Shukla et al., 2018; Jeon and Kim, 2019; Li et al., 2019). The criteria applied are (1) ‘normal SSR’ as a conventional definition from monoSSR to hexaSSR, (2) ‘extented SSR’ referring from heptaSSR (repeats of 7 bp unit) to decaSSR (repeats of 10 bp unit), and (3) ‘potential SSR’ referring specific cases with only 2 units in pentaSSR and hexaSSR. These criteria have been applied and provided better understanding of SSR patterns in previous analyses in chloroplast genomes of Dysphania species (Kim et al., 2019), Arabidopsis thaliana (L.) Heynh. (Park et al., 2020a), Chenopodium album L. (Park et al., 2021a), and mitochondrial genome of Rosa rugosa Thunb. (Park et al., 2020b).
Nucleotide diversity analysis of chloroplast genomes
We calculated nucleotide diversity in the full chloroplast genomes from eight taxa of the genus Daphne and Diarthron linifolium using the method proposed by Nei and Li (Nei and Li, 1979) with the perl script. Nucleotide diversities were scanned along the genome with 500-bp window size and 200-bp step size for overlapped sliding windows. Our script for nucleotide diversity calculation has been utilized in previous studies (Kim et al., 2019; Lee et al., 2020; Park et al., 2020a, 2020c, 2021a; Lee and Park, 2021; Park and Xi, 2021).
Construction of phylogenetic trees
Whole chloroplast genomes of 31 Thymelaeaceae including one outgroup species, Aquilaria rostrata (Hishamuddin et al., 2020), were aligned using MAFFT v7.450 software (Katoh and Standley, 2013). The maximum-likelihood (ML) and Neighbor-joining (NJ) trees were inferred in MEGA X (Kumar et al., 2018) using heuristic search with Nearest Neighbor Interchange (NNI) branch swapping, Tamura-Nei model, and uniform rates among sites. All other options were set as default. We performed bootstrap analyses with 1,000 and 10,000 pseudoreplicates for ML and NJ methods, respectively, with the same option. Posterior probability of each node was estimated by Bayesian inference using MrBayes v3.2.6 (Huelsenbeck and Ronquist, 2001) implemented in the software package, Geneious Prime v2020.2.4. The HKY85 model with gamma rates was used as a molecular model. A Markov chain Monte Carlo algorithm was employed for 1,100,000 generations with four chains running simultaneously. To build a consensus tree, we sampled trees every 200 generations after removing trees from the first 100,000 generations as burn-in.
Results and Discussion
Complete chloroplast genome of Diarthron linifolium
Chloroplast genome of D. linifolium is 172,644 bp and has four subregions: 86,158 bp of large single copy (LSC), 2,858 bp of small sing copy (SSC) regions, and two inverted repeat regions (IR; 41,814 bp) separating LSC and SSC in circular genome structure (Fig. 1). In the LSC and SSC regions, we identified 138 genes (92 protein-coding genes, eight rRNAs, and 38 tRNAs) while 28 genes (16 protein-coding genes, eight tRNAs, and four rRNAs) are duplicated in two IR regions (Fig. 1). The SSC region of Diarthron linifolium chloroplast genome is extremely short, which is also found in the chloroplast genomes of neighbor species, including Daphne species (Cho et al., 2018; Yan et al., 2019a, 2019b, 2021) except Daphne genkwa Siebold & Zucc. (Yoo et al., 2021). The overall GC content is 36.3% and those in the LSC, SSC, and IR regions are 35.0%, 29.3%, and 38.9%, respectively. Chloroplast genome sequence can be accessed via accession number MW566785 in GenBank of NCBI at https://www.ncbi.nlm.nih.gov. The associated BioProject, SRA, and Bio-Sample numbers are PRJNA692680, SRR13485453, and SAMN17360683, respectively.

Circular map, simple sequence repeat analysis, and nucleotide diversity analysis of Diarthron linifolium complete chloroplast genome. A. The genes located outside of the circle are transcribed clockwise, while those located inside are transcribed counterclockwise. The dark gray plot in the inner circle corresponds to GC content. Large single copy (LSC), small single copy (SSC), and inverted repeat (IR) are indicated with LSC, SSC, and IR (IRA and IRB), respectively. B. X-axis presents simple sequence repeat (SSR) types based on motif length and Y-axis shows frequency of SSRs. Based on SSR types, different colors were used along with normal SSRs, potential SSRs, and extended SSRs. Each bar contains two parts: genic SSRs displayed in the bottom of bars and intergenic SSRs occupied in the upper part of each bar. C. X-axis shows chloroplast coordination and Y-axis presented nucleotide diversity calculated based on the sliding window method (See “Materials and Methods”). Blue graph indicates nucleotide diversity along with chloroplast genomic coordination and black labels on the peak indicate position of peaks based on gene names around them. Blue-colored arrows indicate LSC and SSC regions and orange-colored arrows mean IR regions.
Identification of SSRs on Diarthron linifolium chloroplast genomes
SSRs have been used for developing molecular markers (Huang et al., 2015; Li et al., 2020a, 2020b). We identified 90 SSRs as normal SSRs, 542 potential SSRs, and 63 extended SSRs from the analysis using SSRDB pipeline (See “Materials and Methods”) (Fig. 1B, Supplementary Table 2). Our results indicated that the number of identified SSRs from D. linifolium is higher than those identified in Chenopodium album (Park et al., 2021a), Arabidopsis thaliana (Park et al., 2020a) and four Dysphania species (Kim et al., 2019). This result can be explained that the chloroplast genome size of D. linifolium is larger than those in aforementioned species. In the case of the Plantago depressa Willd. (Kwon et al., 2019), and Plantago fengdouensis (Z. E. Chao & Yong Wang) Yong Wang & Z. Yu Li (Wang et al., 2020) chloroplast genomes, of which length is similar to D. linifolium, numbers of normal and potential SSRs are still smaller (Park et al., unpubl. data), suggesting that D. linifolium chloroplast genome contains relatively large number of SSRs.
In detail, monoSSR displayed the high proportion (71.11%; 64 monoSSRs) among normal SSRs and HeptaSSR exhibited the highest among extended SSRs (66.67%; 42 HeptaSSRs) (Fig. 1B). This trend of numbers of normal and extended SSRs was also found in all four species. Seventeen genic normal (18.88%), 171 genic potential (31.55%), and 13 genic extended SSRs (20.63%) were identified (Fig. 1B), displaying the different proportion of genic SSRs along with three types of SSRs. Among them, ycf1 and ycf2 contained the largest number of SSRs (46 and 36, respectively) (Supplementary Fig. S1). These SSRs may have more intraspecific or interspecific variations because these genes displayed high nucleotide diversities in various plant species (Hong et al., 2017; Jiang et al., 2017; Liu et al., 2018; de Souza et al., 2019; Kim et al., 2019; Li et al., 2019; Park et al., 2020c, 2021a; Loeuille et al., 2021), which can be considered as molecular markers to distinguish species or intraspecific taxa (Neubig et al., 2009; Dong et al., 2015). The remaining genic SSRs will not have many variations in comparison to those in ycf1 and ycf2, which will be utilized as molecular markers to recognize relatively high level of taxa.
Nucleotide diversity analysis of Diarthron linifolium and Daphne chloroplast genomes
Using eight Daphne and Diarthron linifolium chloroplast genomes, the nucleotide diversity was investigated to understand variable regions on chloroplast genomes. We excluded two Daphne genkwa chloroplast genomes because it was clustered into the group consisting of Wikstroemia species (Yoo et al., 2021). Overall nucleotide diversity was 0.00244, which is lower than those calculated in four Dysphaina species (0.0068) (Kim et al., 2019) and eight Plantago species (0.01751) (Park et al., unpubl. data) but higher than those from Agrimonia species (0.000727) (Park et al., unpubl. data) and four Viburnum species (0.00176) (Park et al., 2020c).
Eight peaks over 0.01 in nucleotide diversity were identified (Fig. 1C). Seven peaks were found in intergenic regions but one peak in the genic region harboring ndhF (Fig. 1C). The peak in the gene, ndhF was located in the junction between IR and SSC and only two out of nine chloroplast genomes were annotated as ndhF genic region (Supplementary Fig. S2). These regions with high nucleotide diversity can be considered as candidates of molecular markers to distinguish species of Daphne and Diarthron.
Phylogenetic analysis of Thymelaeaceae chloroplast genomes
Phylogenetic tree exhibited that Diarthron linifolium was placed as a sister of the group composed of species in the genus Daphne (Fig. 2). Our results of genome data are consistent with previous analyses of a few chloroplast regions such as rbcL (Van der Bank et al., 2002; Beaumont et al., 2009) and whole plastid genomes (Yoo et al., 2003; He et al., 2021). Although more species of Diarthron should be included in further phylogenetic analysis, the current chloroplast genome data suggest that Diarthron is distantly related with Stellera. Similarity between Diarthron and Stellera in gross morphology, both are adapted to dry environment, may have derived independently. Diarthron is easily distinguished from Stellera by having 4-merous flowers, while Stellera has 5- or 6-merous flowers.
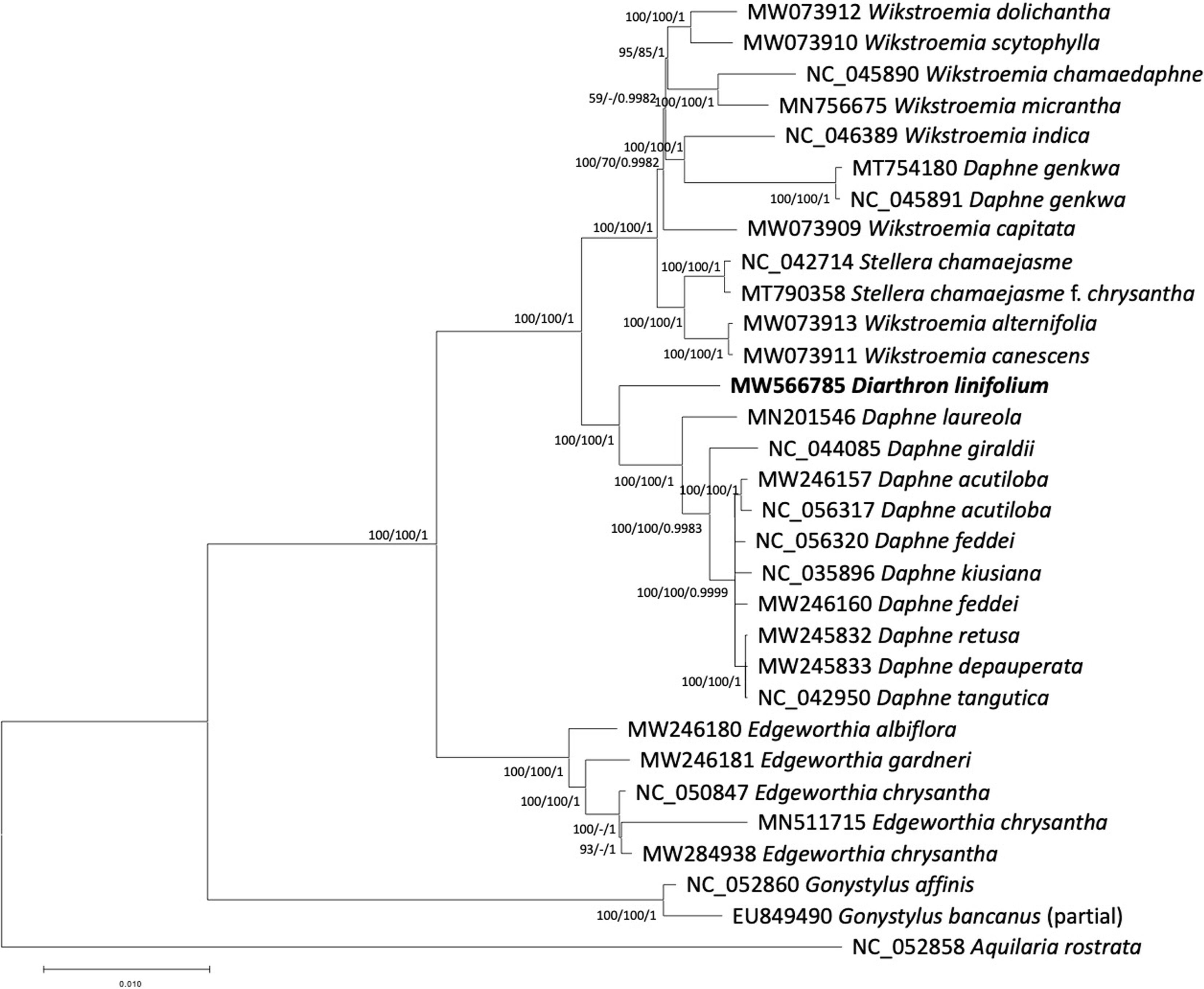
Phylogenetic tree inferred from 31 chloroplast genomes in Thymelaeaceae. Maximum-Likelihood tree is presented. The numbers above branches indicate bootstrap support values of maximum-likelihood (1,000 bootstrap repeats) and Neighbor-joining methods (10,000 bootstrap repeats), and posterior probabilities from Bayesian inference.
Supplementary Materials
Online supplemental data can be found at https://www.e-kjpt.org.
List of the 31 chloroplast genomes used in this study.
List of the SSRs identified on the D. linifolium chloroplast genome
Number of simple sequence repeats (SSRs) located in genic regions along with protein-coding genes (PCGs)
Nucleotide alignment of the nine Daphne and Diarthron chloroplast genomes around the small single copy (SSC) region.
Acknowledgements
We are grateful to Hye Ryun Na for the help of fieldwork and Yun-Gyeong Choi for the laboratory assistance. This work was supported by The Catholic University of Korea under the new faculty support grant (No. M-2020-B0014-00004) to S-TK.
Notes
Conflicts of Interest
Sang-Hun OH, a contributing editor of the Korean Journal of Plant Taxonomy, was not involved in the editorial evaluation or decision to publish this article. All remaining authors have declared no conflicts of interest.