The complete mitochondrial genome of Arabidopsis thaliana (Brassicaceae) isolated in Korea
Article information

Abstract
Arabidopsis thaliana (L.) Heynh. is a small plant species that serves as a model organism of plant biology and genetics. Here, we present the first complete mitochondrial genome of Korean A. thaliana natural isolate (named as 180404IB4), which is 368,875 bp long and contains 58 genes (33 protein-coding genes, 22 tRNAs, and three rRNAs), with a GC ratio of 44.8%. Sixty-four single-nucleotide polymorphisms and 11 insertion and deletion regions (1,089 bp in length) are identified against the Col-0 ecotype, showing one large insertion of 1,069 bp without structural variation. Phylogenetic trees constructed from 30 conserved genes indicate that the 180404IB4 mitochondrial genome is clustered with Col-0 and three East Asian ecotypes.
Arabidopsis thaliana (L.) Heynh. is a small plant species distributed in Eurasia and Africa (Hoffmann, 2002). It is a model organism of plant biology and genetics (Rensink and Buell, 2004) because of its rapid life cycle (ca. 10 weeks) (Mandel and Yanofsky, 1995) and relatively small genome size (ca. 119 Mb) (The Arabidopsis Genome Initiative, 2000). As next-generation sequencing technologies have been established, above one thousand whole genomes have been sequenced (Gan et al., 2011; Schmitz et al., 2013): e.g., 1,135 natural isolates from Eurasian and North African (The 1001 Genomes Consortium, 2016) and 118 strains from Yangtze River in China (Zou et al., 2017). In contrast, only six complete chloroplast (Park et al., 2020a) and three mitochondrial genomes (mitogenomes) (Unseld et al., 1997; Davila et al., 2011; Sloan et al., 2018) are available even though they are valuable to investigate phylogenetic relationship (Park et al., 2020a, 2020c, 2021). Here, we presented the complete mitogenome of A. thaliana isolated in Korea together with three East Asian isolates to understand their phylogenetic relationship.
Materials and Methods
Plant material
We collected an individual accession of A. thaliana in the population located in Yeonggwang-gun, Jeollanam-do province in Korea (180404IB4). A voucher and isolated DNA were deposited in the InfoBoss Cyber Herbarium (IN, the voucher number IB-00925).
DNA extraction and mitochondrial genome determination
Its total DNA was extracted from fresh leaves by using a DNeasy Plant Mini Kit (QIAGEN, Hilden, Germany). Genome sequencing was performed using HiSeqX at Macrogen Inc., Korea, and de novo assembly was done by Velvet v1.2.10 (Zerbino and Birney, 2008) and GapCloser v1.12 (Zhao et al., 2011). Assembled sequences were modified and confirmed by BWA v0.7.17 (Li, 2013) and SAMtools v1.9 (Li et al., 2009). All bioinformatic analyses were conducted in the Genome Information System (http://geis.infoboss.co.kr/) like the previous studies (Kwon et al., 2019a, 2019b; Park et al., 2019a, 2019d, 2020b; Min et al., 2020; Choi et al., 2021a, 2021b; Kim et al., 2021). The same method was applied to assemble mitogenomes of three East Asian A. thaliana isolates based on public raw reads (Table 1).
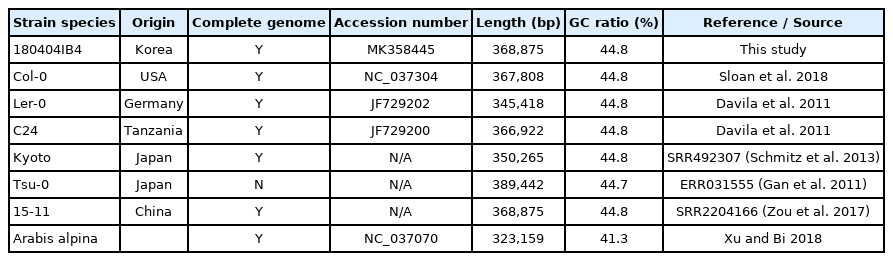
List of the mitochondrial genomes used in this study
Genome annotation was conducted based on Col-0 mitogenome (NC_037304) (Sloan et al., 2018) with Geneious Prime 2020.2.4 (Biomatters Ltd., Auckland, New Zealand). A circular map of A. thaliana mitogenome was drawn using OGDRAW v1.31 (Greiner et al., 2019).
Phylogenetic analysis
Maximum-Likelihood (ML) and Bayesian inference (BI) phylogenetic trees were constructed based on the concatenated alignment of the 30 aligned conserved genes from the seven A. thaliana including Korean individual and three ecotypes (Table 1) and Arabis alpina (Xu and Bi, 2018) mitogenomes (Table 1) by MAFFT v7.450 (Katoh and Standley, 2013). The ML tree was reconstructed in MEGA X with 1,000 bootstrap repeats (Kumar et al., 2018). In the ML analysis, a heuristic search was used with nearest-neighbor interchange branch swapping, TVM + F + R4 model determined by jModelTest v2.1.5 (Darriba et al., 2012), and uniform rates among sites. All other options used the default settings. The posterior probability of each node was estimated by BI using MrBayes v3.2.6 (Huelsenbeck and Ronquist, 2001) plug-in implemented in Geneious Prime 2020.2.4 (Biomatters Ltd.). The HKY85 model with gamma rates was used as a molecular model. A Markov chain Monte Carlo algorithm was employed for 1,100,000 generations, sampling trees every 200 generations, with four chains running simultaneously. Trees from the first 100,000 generations were discarded as burn-in.
Data availability
Mitochondrial genome sequence of Korean A. thaliana can be accessed via accession number MK358445 in NCBI GenBank. The associated BioProject, SRA, and Bio-Sample numbers are PRJNA727818, SAMN19040823, and SRR14458670, respectively.
Results and Discussion
The complete mitogenome of A. thaliana 180404IB4 (MK358445) is 368,875 bp long (Fig. 1A), longer than three previously assembled and one newly assembled A. thaliana mitogenomes (Table 1). Overall GC content is 44.8% and it contains 58 genes (33 protein-coding genes, 22 tRNAs, and three rRNAs) (Fig. 1A), which are the same as the Col-0 mitogenome.
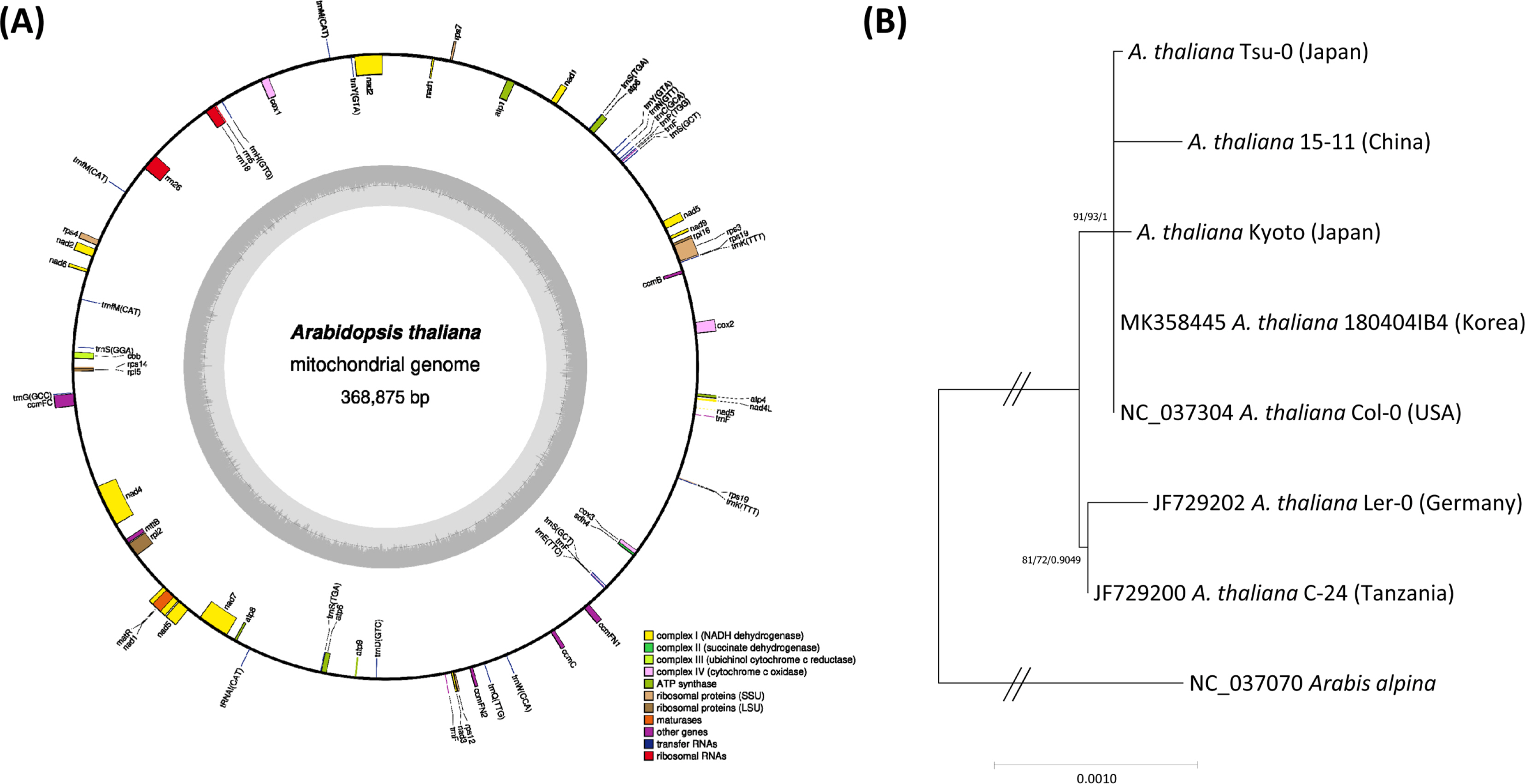
A. Circular map of mitochondrial genome of Arabidopsis thaliana isolated in Korea. Gene shown outside are transcribed clockwise, and inside the circle are transcribed counter clockwise. Genes are colour-coded to distinguish different functional groups. The dark grey and the light grey plot in the inner circle correspond to the GC content and AT content, respectively. B. Maximum-Likelihood (ML) and Bayesian inference (BI) phylogenetic trees of 30 conserved genes from seven Arabidopsis and one Arabis complete mitochondrial genomes. Phylogenetic tree was drawn based on ML tree. The numbers above branches indicate bootstrap support value of ML tree and posterior probability of BI tree, respectively.
Sixty-four single nucleotide polymorphisms (SNPs; 0.017%) and 11 insertion and deletion (INDEL) regions (1,089 bp in length; 0.30%) were identified against Col-0 mitogenome. Interestingly, one 1,069-bp insertion was found, similar to the insertions identified in chloroplast genomes of Coffea arabica L. (Park et al., 2019c) and Duchesnea chrysantha (Zoll. & Moritzi) Miq. (Park et al., 2019b) and mitogenomes of Liriodendron tulipifera L. (Park et al., 2019a) and Populus alba x Populus glandulosa (Park et al., 2019d). Proportion of intraspecific variations identified between 180404IB4 and Col-0 is similar to that of Rosa rugosa Thunb. (124 SNPs, 0.041% and 769-bp INDELs, 0.25%) (Park et al., 2020b), is less than those of Liriodendron tulipifera L. (365 SNPs, 0.07% and 2,117-bp INDELs, 0.38%) (Park et al., 2019a) and Glycyrrhiza uralensis Fisch. ex DC. (1,099 SNPs, 0.24% and 1,736-bp INDELs, 0.38%) (Baek et al., under review), but is greater in number than that of Malus x domestica Borkh. (140 SNPs, 0.035% and 6-bp INDELs, 0.0015%) (Goremykin et al., 2012; Ge et al., 2020).
No structural variation among 180404IB4, 15-11, and Col-0 mitogenomes was found, while the remaining A. thaliana genomes (Table 1) presented structural variations (Davila et al., 2011). It is also congruent to the recent analysis which presents a dynamic structure of A. thaliana mitogenome along with different ecotypes (Masutani et al., 2021).
Phylogenetic trees constructed from the conserved genes of the seven A. thaliana and Arabis alpina mitogenomes (Table 1) show that four East Asian Arabidopsis including 180404IB4 were clustered with the Col-0 (USA) with high supportive values and Ler-0 (Germany) and C-24 (Tanzania) were clustered in an independent clade (Fig. 1B). It is different from the phylogenetic tree constructed from the complete chloroplast genomes, displaying the intraspecific topology of the six A. thaliana ecotypes except for C-24 with high supportive values (Park et al., 2020a). It might be caused by the low coverage of the 30 conserved genes (6.98%) against the complete mitogenome in comparison to that of the chloroplast genomes. To improve the resolution of phylogenetic analysis using mitogenomes, more conserved areas except genic regions should be rescued and addition mitogenomes of East Asian A. thaliana are needed to be assembled to investigate their phylogenetic relationship. Throughout this study, this mitogenome presents that Korean A. thaliana is phylogenetically related to the other East Asian isolates and Col-0.
Acknowledgements
This work was supported by InfoBoss Research Grant (IBG-0023).
Notes
Conflict of Interest
The authors declare that there are no conflicts of interest.