A report of the second chloroplast genome sequence in Veronica nakaiana (Plantaginaceae), an endemic species in Korea
Article information

Abstract
Veronica nakaiana Ohwi (Plantaginaceae) is an endemic taxon on Ulleungdo Island, Korea. We report the second complete chloroplast genome sequence of V. nakaiana. Its genome size is 152,319 bp in length, comprising a large single-copy of 83,195 bp, a small single-copy of 17,702 bp, and a pair of inverted repeat regions of 25,711 bp. The complete genome contains 115 genes, including 51 protein-coding genes, four rRNA genes, and 31 tRNA genes. When comparing the two chloroplast genomes of V. nakaiana, 11 variable sites are recognized: seven SNPs and four indels. Two substitutions in the coding regions are recognized: rpoC2 (synonymous substitution) and rpl22 (nonsynonymous substitution). In nine noncoding regions, one is in the tRNA gene (trnK-UUU), one is in the intron of atpF, and seven are in the intergenic spacers (trnH-GUG~psbA, trnK-UUU, rps16~trnQ-UUG, trnC-GCA~petN, psbZ~trnG-GCC, ycf3~trnS-GGA, ycf4~cemA, and psbB~psbT). The data provide the level of genetic variation in V. nakaiana. This result will be a useful resource to formulate conservation strategies for V. nakaiana, which is a rare endemic species in Korea.
Veronica L. (Plantaginaceae) contains 450 species worldwide and includes many economically important species for medicinal purposes (Salehi et al., 2019). Its diversification centers are western Asia and New Zealand (Albach and Meudt, 2010). Psudolysimachion has been proposed by Opiz (1852) based on section Pseudolysimachium W. D. J. Koch of Veronica. This taxon has been recognized as an independent genus in previous Korean and Japanese publications (Yamazaki, 1968; Lee and Yamazaki, 1983) based on unique characters compare to other Veronica, such as basal chromosome number (x = 17), long corolla tube, and densely packed inflorescences (Albach et al., 2004a, b). However, recent molecular studies confirmed that members of Pseudolysimachion were included in the clade of Veronica (Albach et al., 2004a, b; Albach and Meudt, 2010). Therefore, this taxon is currently recognized as a subgenus in the classification system of Veronica, which contains 11 subgenera (Albach, 2004a, b; Albach, 2008).
On the Korean peninsula, eight species and 14 taxa have been reported in the subgenus Pseudolysimachium of Veronica (Kim and Choi, 2007). As an endemic species in Korea, Veronica nakaiana Ohwi is distributed only on Ulleungdo Island (Kim, 2018; recognized as Pseudolysimachion nakaianum (Ohwi) T. Yamaz.). Many studies on endemic species of Ulleungdo Island have been conducted to understand the evolution of genetic diversification of the species on Ulleungdo Island of volcanic origin. However, all of these studies are based on sequence data from a few DNA regions (e.g., Jeong et al., 2014; Oh, 2015; Lee et al., 2017; Cheong et al., 2020), often resulting in the lack of molecular variations within the species. Therefore, studies based on the whole chloroplast (cp) genome are needed in future population genetic studies of endemic species (Park et al., 2020).
The first cp genome sequence of V. nakaiana has been reported along with other related species of Veronica (Choi et al., 2016). In this study, we report the second cp genome of V. nakaiana from the sample collected in a different location from Choi et al. (2016) to recognize the infraspecific variation of cp genome. Our results, based on the entire organelle genome level, will provide important information for the understanding of the evolution and genetic diversification of this species.
Materials and Methods
Plant material
We collected a plant from Ulleungdo Island (37°63′17.56″N, 127°02′66.88″E) in 2017, and the plant has been cultivated in the greenhouse of the Sungshin University. A voucher specimen for this study is prepared with a part of the plant of which leaves were used for the DNA extraction and is deposited in the herbarium of the Sungshin University (voucher number: Y.-E. Lee 2020-001, SWU).
Sample preparation and cp genome determination
Total genomic DNA was extracted from leaves using a commercial kit (GeneAll Plant SV Mini Kit, GeneAll Biotechnology Co. Ltd., Seoul, Korea). We conducted the Next Generation Sequencing based on the MGISEQ platform (100 bp paired-end reads; MGI Tech Co. Ltd., Shenzhen, China).
We used the previously reported cp genome sequence of the V. nakaiana (NC_031153) as a reference sequence to assemble the new cp genome sequence. We mapped each paired-end read against a reference using “Geneious” module included in the Geneious program (v9.0.5) (Kearse et al., 2012) with the ‘medium-low’ sensitivity option.
A consensus sequence was produced after checking the borders of the inverted repeat regions (IRs). Six specific primers located in rps19, rpl2, ycf1, ndhF, and trnH-GUG were designed to confirm the low-covered-read parts around IR borders (Table 1). The PCR has conducted with a total volume of 20 μL containing 10 μL Master Taq (2× PCR Master-mix Solution i-Taq, iNtRON, Seoul, Korea), 1 μL of each primer, 7 μL of distilled water, and 1 μL template DNA (10 ng/μL). The PCR was conducted using S1000 Thermal cycler (Bio-Rad, Hercules, CA, USA) with the following file: 3 min predenaturation (94°C) followed by 35 cycles of 30 s of denaturation (95°C), 30 s of annealing (55°C), and 45 s of extension (72°C), and finished by a final extension of 7 min (72°C). The PCR products were checked in 1.3% agarose gel with 0.001% ethidium bromide under the UV light using the Gel Doc XR+ System (Bio-Rad). Bidirectional Sanger sequencing was performed by a 3730xl DNA analyzer (Macrogen Inc., Seoul, Korea) with the same primer sets that we designed for PCR in each PCR product. Sequences were assembled and edited using Sequencher 4.9 (Gene Code Corporation, Ann Arbor, MI, USA). Consensus sequences from the Sanger sequencing were compared with the consensus sequence from the next generation sequencing (NGS) data. We performed the gene annotation using Geneious (v.9.0.5) (Kearse et al., 2012) based on a reference genome, the first cp genome from V. nakaiana (NC_031153) (Choi et al., 2016). We drew a circular map of the cp genome using OGdraw (ver. 1.3.1) (Greiner et al., 2019).

Primers for PCR and Sanger sequencing used in this study.
Phylogenetic analysis
The phylogenetic analysis has performed with ten cp genomes of Plantaginaceae deposited on the organellar genome databases of the NCBI to date including a cp genome determined in this study (Veronica: NC_031153, NC_031344, and MT422349; Veronicastrum: NC_031345; Plantago: NC_041161, NC_028520, NC_028519, NC_041421, and NC_041420; Digitalis: NC_034688) and an outgroup taxon from Scrophulariaceae (Hemiphragma: NC_045398). The outgroup taxon is selected based on a recent phylogenetic study of the Lamiales (Refulio-Rodriguez and Olmstead, 2014). The full-length sequences of these genomes are aligned using MAFFT (v7.308) (Katoh and Standley, 2013) module in the Geneious (v9.0.5) (Kearse et al., 2012). We selected GTR + I + Γ as the best base-substitution model using the Modeltest module (Posada and Crandall, 1998) in MEGA7 (Kumar et al., 2016). The maximum-likelihood analysis is performed using raxmlGUI (ver. 1.5) (Silvestro and Michalak, 2012) with 1,000 bootstrap replications.
Results and Discussion
We obtained 39,341,301 reads (11.8 Gbp) after the quality filtration using the fastQC (v0.11.8; www.bioinformatics.babraham.ac.uk/projects/fastqc/). A total of 2,817,887 reads were mapped to the reference sequence, which was 7.16% of the total sequence obtained.
The consensus sequence of mapped reads showed an almost perfect match with a reference (NC_031153) except for 11 different sites (Table 2). Four border regions of IRs, which showed a low level of read-coverage, were compared with the Sanger sequences from PCR products based on specific primers (Table 1). The Sanger sequences were perfectly matched with the consensus sequence from the NGS.

Sequence variation between two chloroplast genomes of Veronica nakaiana.
The second complete cp genome of V. nakaiana determined in this study (GenBank accession: MT422349) has 152,319 bp in length (GC ratio is 37.9%) and composed of four subregions: 83,195 bp of large single-copy (LSC; 54.62%), 17,702 bp of small single-copy (11.62%) regions, and 25,711 bp of a pair of IRs (33.76%) (Fig. 1). The genome includes 115 genes comprising 80 protein-coding genes, four rRNA genes, and 31 tRNA genes.
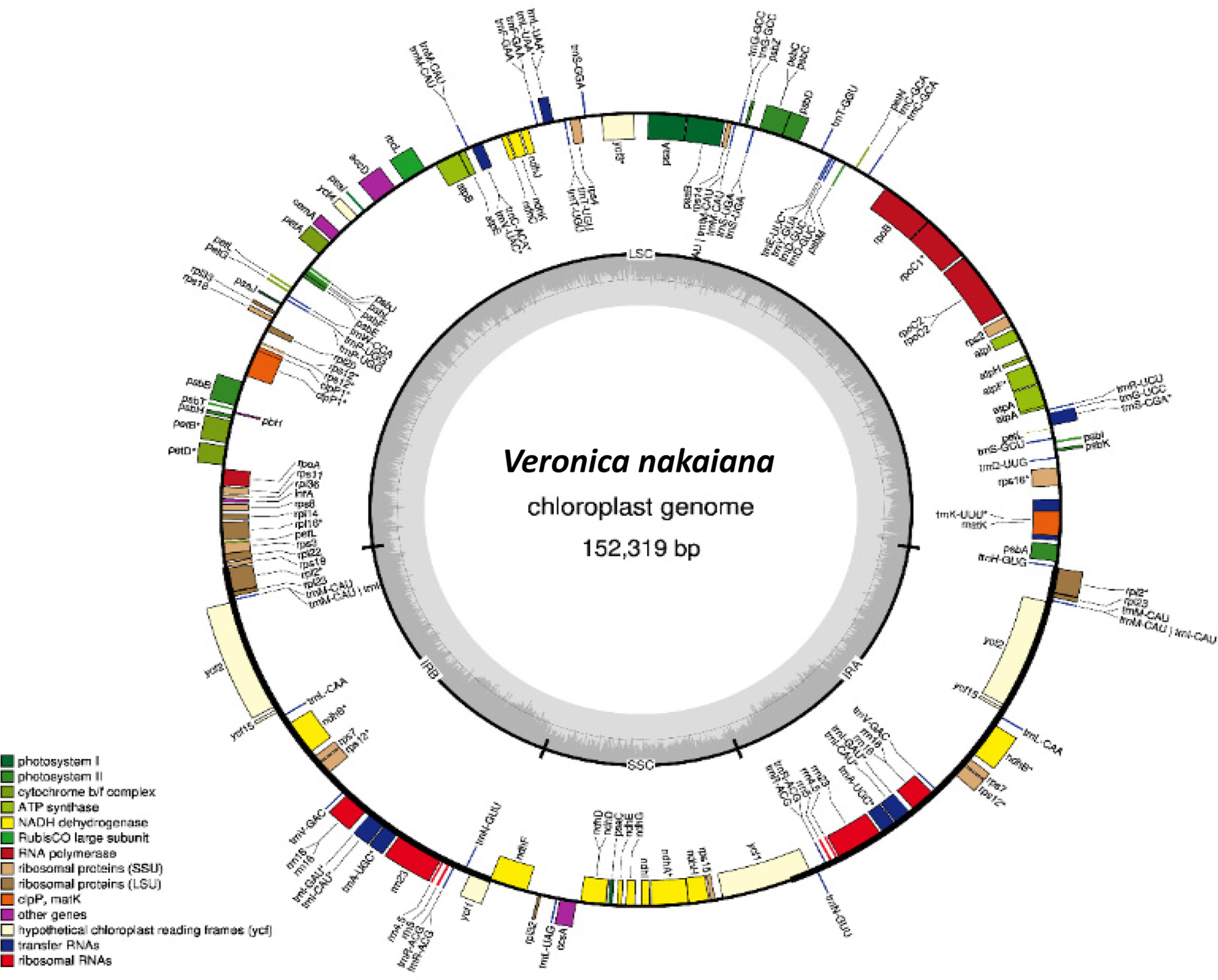
Structural map of the chloroplast genome in Veronica nakaiana. Asterisks indicate genes containing intron.
By comparing with the first cp genome of V. nakaiana (Choi et al., 2016), 11 different sequences, including four indels, are identified. All of them are found in the LSC region: seven of them (trnH-GUG–psbA, trnK-UUU, rps16–trnQ-UUG, trnC-GCA– petN, psbZ–trnG-GCC, ycf3–trnS-GGA, ycf4–cemA, and psbB–psbT) are in the intergenic spacers, two are in the coding regions (rpoC2 and rpl22), one is in tRNA (trnK-UUU), and one is in the intron of atpF. In the two protein-coding genes, a change (T/G) in the 3,861st nucleotide of rpoC2 located at the third codon position is a synonymous substitution. Another change (T/C) in the 61st nucleotide of rpl22 located at the first codon position is a nonsynonymous substitution that results in a change from cysteine to arginine.
The maximum-likelihood tree shows that two cp genomes from V. nakaiana form a clade, and this clade is a sister to V. persica which is another cp genome from Veronica (Fig. 2). Veronicastrum sibricum, a representative of Veronicastrum, is a sister to a monophyletic Veronica clade.
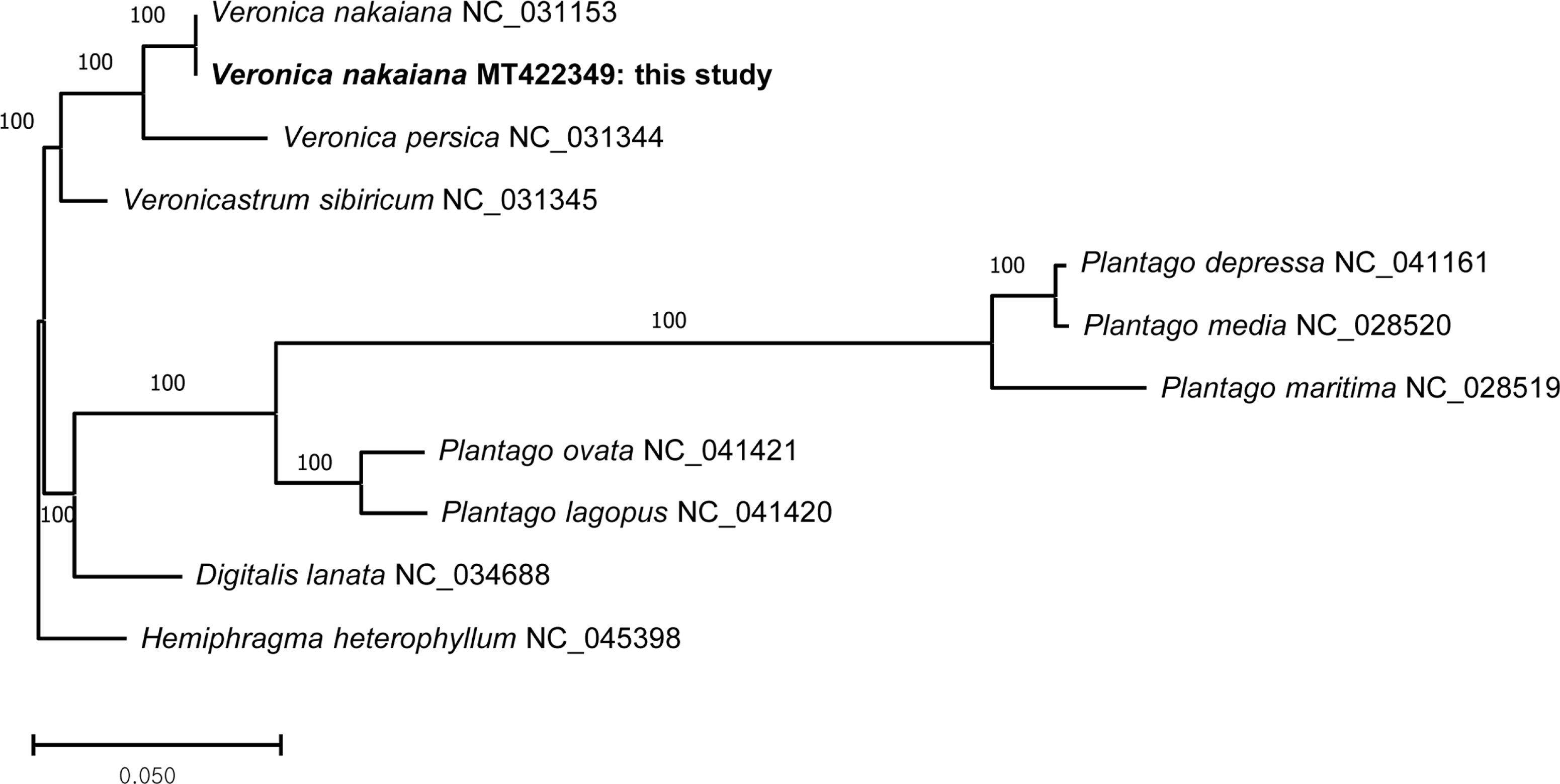
A maximum-likelihood tree using ten selected chloroplast genomes from Plantaginaceae, including a genome determined in this study and an outgroup (Scrophulariaceae). The numbers above the node indicate bootstrap values.
Haplotype diversity of several endemic plants on Ulleungdo Island have been studied based on a small number of DNA regions. Variable haplotypes in cpDNA regions were recognized in Campanula takesimana Nakai (rps16-trnK, trnQ-rps16, psbD-trnT, and psbM-trnD) (Cheong et al., 2020) and Rubus takesimensis Nakai (trnL-trnF) (Lee et al., 2017). In contrast, no haplotype variations were reported in the selected cp DNA regions for Lonicera insularis Nakai (trnL-trnF, trnS-trnG, psbM-trnD, and matK) (Jeong et al., 2014) and Fagus multinervis Nakai (trnH-psbA) (Oh, 2015).
Analysis of sequence variation between two cp genomes in V. nakaiana in this study is a useful resource to understand the evolution and diversification of this species. This study will also contribute to the conservation and propagation studies in V. nakaiana, which is a rare endemic species in Korea.
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF-2015R1D1A1A02062467) to S. Kim.
Notes
Conflict of Interest
The authors declare that there are no conflicts of interest.