Chromosome numbers and polyploidy events in Korean non-commelinids monocots: A contribution to plant systematics
Article information

Abstract
The evolution of chromosome numbers and the karyotype structure is a prominent feature of plant genomes contributing to or at least accompanying plant diversification and eventually leading to speciation. Polyploidy, the multiplication of whole chromosome sets, is widespread and ploidy-level variation is frequent at all taxonomic levels, including species and populations, in angiosperms. Analyses of chromosome numbers and ploidy levels of 252 taxa of Korean non-commelinid monocots indicated that diploids (ca. 44%) and tetraploids (ca. 14%) prevail, with fewer triploids (ca. 6%), pentaploids (ca. 2%), and hexaploids (ca. 4%) being found. The range of genome sizes of the analyzed taxa (0.3–44.5 pg/1C) falls well within that reported in the Plant DNA C-values database (0.061–152.33 pg/1C). Analyses of karyotype features in angiosperm often involve, in addition to chromosome numbers and genome sizes, mapping of selected repetitive DNAs in chromosomes. All of these data when interpreted in a phylogenetic context allow for the addressing of evolutionary questions concerning the large-scale evolution of the genomes as well as the evolution of individual repeat types, especially ribosomal DNAs (5S and 35S rDNAs), and other tandem and dispersed repeats that can be identified in any plant genome at a relatively low cost using next-generation sequencing technologies. The present work investigates chromosome numbers (n or 2n), base chromosome numbers (x), ploidy levels, rDNA loci numbers, and genome size data to gain insight into the incidence, evolution and significance of polyploidy in Korean monocots.
Chromosome numbers and karyotype structure have always been considered to be an important character in analyses of the phylogenetic relationships and evolutionary processes in angiosperms (Levin and Wilson, 1976; Guerra, 2008; Jang et al., 2013). To date, chromosome numbers have been reported for about 25–30% of flowering plants (Bennett, 1998; Weiss-Schneeweiss and Schneeweiss, 2013). The chromosome numbers in angiosperms vary 160–fold (Weiss-Schneeweiss and Schneeweiss, 2013) ranging from 2n = 4 (Poaceae, Hyacinthaceae, Asteraceae, Cyperaceae: Vanzela et al., 1996; Roberto, 2005) to 2n = 640 (Crassulaceae: Uhl, 1978). The haploid chromosome numbers of the majority of angiosperms range between n = 7 and n = 20 (Grant, 1982; Masterson, 1994). Taxonomic groups display varying degrees of chromosome number changes both among and within genera (e.g., 2n = 8, 10, 12, 14, 19, 20, 25, 26, 27, 28, 35, 42 in Prospero/Hyacinthaceae: Jang, 2013; 2n = 18, 20, 22, 24, 28, 36, 40, 46, 48, 54, 56, 60, 66 in Melampodium/Asteraceae: Stuessy, 1971; Weiss-Schneeweiss et al., 2009; 2n = 24 in Lilium/Liliaceae: Sultana et al., 2010), and such changes continue to be used in systematics and elucidating evolutionary patterns within these groups of plants (Mayrose et al., 2010; Schubert and Lysak, 2011; Husband et al., 2013; McCann et al., 2016).
Hybridization and polyploidization have been commonly observed in many economically important plant groups (Lim et al., 2007; Mandáková et al., 2013), but recent studies have demonstrated that these processes have also been a major force in the diversification and speciation of angiosperms in general (Leitch and Leitch, 2008). Hybrids and polyploids experience numerous chromosomal rearrangements (e.g., inversions, deletions, translocations, centromeric shifts, etc.) and more subtle changes in sequence composition (sequence loss or gain, expansion/reduction of repetitive DNA), and they continue to generate species diversity contributing to speciation events (Soltis and Soltis, 2009; Weiss-Schneeweiss and Schneeweiss, 2013). The propensity for polyploidization appears to be unequally distributed in plant groups with polyploidy in angiosperms being more common in monocots (ca. 58%) than in dicots (ca. 43%) (Soltis and Soltis, 2009; Weiss-Schneeweiss et al., 2013).
There are two general types of polyploidy: autopolyploidy (i.e., multiplication of chromosome sets within a single species or genome) and allopolyploidy (i.e., multiplication of chromosome sets accompanied by merger of genomes of two or more species), both of which arise as a result of a failure of either meiotic or mitotic cell division (Stebbins, 1971; Otto and Whitton, 2000; Ramsey and Schemske, 2002). Although autopolyploidy has historically been considered as less frequent and less important than allopolyploidy (Stebbins, 1971; Soltis et al., 2007), natural autopolyploids are much more common than originally assumed (Ramsey and Schemske, 2002; Parisod et al., 2010), as recent studies continue to demonstrate. Multiple ploidy levels have been demonstrated to exist within many species (autopolyploidy), which often influences the degree of morphological variation in those taxa. Current focus of polyploidy research is on the genetic, epigenetic, chromosomal, and genomic consequences of polyploidization (Bowers et al., 2003; Liu and Wendel, 2003; Osborn et al., 2003; Rapp and Wendel, 2005), mechanisms of polyploid formation and establishment (Ramsey and Schemske, 2002), the ecological effects of polyploidization (Weiss-Schneeweiss et al., 2013; Soltis et al., 2016), and most of all, the impact of polyploidy on plant diversity (Mandáková et al., 2017; Jang et al., 2018).
Modern cytology greatly profits from technical advances especially in situ hybridization (e.g., fluorescence in situ hybridization [FISH] and genomic in situ hybridization [GISH], respectively), large scale screening for polyploidy incidence using flow cytometry, and the advent of next-generation sequencing (NGS) technologies. These allow identification, quantification and localization on the genomes of various repeat types, which contribute to genome size variation and changes of which accompany species diversification and speciation (Weiss-Schneeweiss et al., 2015). Repetitive DNA fraction in plant genomes comprises tandem repeats (e.g., satellite DNAs, microsatellites, and ribosomal RNA genes [5S and 35S rRNA genes]) and dispersed repeats represented by mobile genetic elements (Weiss-Schneeweiss et al., 2015). The localization and evolution of tandemly repeated genes encoding 35S (18S-5.8S-25S) and 5S rRNAs in plants have been particularly useful for analysing systematic relationships between closely related species (Weiss-Schneeweiss and Schneeweiss, 2013).
The chromosome numbers in Korean non-Commelinids monocots have previously been reported for a number of taxonomically closely related taxa (Rice et al., 2015, references therein), although the incidence of polyploids and its evolutionary aspects have not been addressed in detail. It is therefore timely to summarize the knowledge of chromosome numbers, genome sizes, and polyploidy incidence in the Korean monocots (Rice et al., 2015; Vitales et al., 2017) and to identify the most important taxonomic groups in which questions of chromosomal evolution can be addressed most effectively.
Chromosome numbers and the incidence of polyploidy in non-commelinids monocot species native to Korea
All available chromosome numbers and base chromosome numbers for Korean non-Commelinids monocots were obtained from the Chromosome Counts Database (CCDB, version 1.45; http://ccdb.tau.ac.il/Angiosperms/, accessed on 2018 May 22) (Rice et al., 2015) following APG IV classification system (Angiosperm Phylogeny Group IV) (Appendix 1) (The Angiosperm Phylogeny Group, 2016). Due to the scarcity of available data on chromosome numbers and ploidy levels variation in Korean Commelinids including Arecales, Commelinales, Poales, and Zingiberales (The Angiosperm Phylogeny Group, 2016), these were excluded from the current analyses.
The systematic ranking of taxa adopted in this study was mainly based on the recent online resources for monocot plants (http://emonocot.org/), the World Checklist of Selected Plant Families (http://wcsp.science.kew.org), the Missouri Botanical Garden Tropicos Database (http://www.tropicos.org/), and the nomenclature was adopted from the most accepted taxonomic treatment for the species based on the Korean Plant Names Index Committee (http://www.nature.go.kr/kpni/index.do) (Appendix 1).
The genome size values and ploidy level inferences in Korean non-Commelinids monocots were retrieved from the Plant DNA C-values database (http://www.kew.org/cvalues/, accessed on 2018 May 22) (Bennett and Leitch, 2012). The data on number and chromosomal localization of rDNA loci (5S and 35S rDNA) in Korean non-Commelinids monocots obtained applying fluorescent in situ hybridization were retrieved from the third release of the plant rDNA database (Vitales et al., 2017; http://www.plantrdnadatabase.com/, accessed on 2018 May 22).
Chromosome numbers are reported for 252 taxa (232 species, 2 subspecies, and 18 varieties) of Korean monocots, with the exception of Commelinids, due to the scarcity of published chromosome numbers for this very speciose this group (Appendix 1). Base chromosome numbers and ploidy levels variation is given for each taxon in Appendix 1. The chromosome numbers reported for Korean non-Commelinids monocots vary between 2n = 2x = 10 in Paris verticillata M. Bieb. and 2n = 40x = 400 in Dioscorea japonica Thunb. (Appendix 1). To date, the documented chromosome numbers in angiosperms vary from 2n = 4 (e.g., Ornithogalum tenuifolium Delaroche in Hyacinthaceae) to 2n = 640 (Sedum suaveolens Kimnach in Crassulaceae), although most species possess between 2n = 14 and 2n = 40 chromosomes (Guerra, 2008; Weiss-Schneeweiss and Schneeweiss, 2013). The base chromosome numbers of analyzed Korean species vary from x = 5 in the genus Paris L. to x = 30 in the genus Hosta Tratt. (Appendix 1). Not only interspecific base chromosome number variation is found in thirteen genera analyzed here (Acorus L., Arisaema Mart., Alisma L., Hydrocharis L., Potamogeton L., Lycoris Herb., Asparagus Tourn. ex L., Polygonatum Mill., Scilla L., Iris Tourn. ex L., Cephalanthera Rich., Gastrodia R. Br., Fritillaria Tourn. ex L.) (Appendix 1) but also intraspecific base chromosome number variation is found within several species (x = 9, 11, 12 in Acorus calmus L.; x = 13, 14 in Arisaema amurense Maxim.; x = 13, 14 in Arisaema peninsulae Nakai; x = 13, 14 in most of taxa in the genus Potamogeton L.; x = 9, 10 in Polygonatum falcatum A. Gray; x = 10, 11 in Polygonatum humile Fisch. ex Maxim.; x = 9, 10, 11 in Polygonatum involucratum (Franch. & Sav.) Maxim.; x = 8, 9 in Scilla scilloides (Lindl.) Druce) (Appendix 1). The incidence of both interspecific (x = 5, 6, 7 in Lotus/Fabaceae: Grant, 1991; x = 9, 10, 11, 12, 13, 14 in Melampodium/Asteraceae: Blöch et al., 2009; x = 3, 4, 5, 6 in Crepis/Asteraceae: Babcock and Jenkins, 1943) and intraspecific base chromosome number variation (x = 5, 6, 7: Prospero autumnale complex: Jang et al., 2013; x = 8, 9: Scilla scilloides complex: Choi et al., 2008) have quite frequently been reported in angiosperms (Husband et al., 2003). Due to very low levels of phenotypic variation and thus lack of diagnostic morphological characters for species delimitations in some taxonomically intricate plant groups (often treated as species complexes), more detailed karyological investigations of the chromosome number variations and karyotype structure are needed for correct interpretation of taxonomic and evolutionary patters as well as classifications of angiosperms in general, but also specifically of monocot species native in Korea in global world-wide context.
Two general types of polyploids can be distinguished, autopolyploids and allopolyploids. Allopolyploids originate via hybridization of at least two different taxa, thus carrying different multiplied sets of chromosomes, while autopolyploids result from multiplication of entire chromosome sets within one taxon, typically species. Thus, both hybridization and polyploidization may play an important role in creating new species diversity in angiosperms (Guerra, 2008; Soltis and Soltis, 2009; Husband et al., 2013; Weiss-Schneeweiss and Schneeweiss, 2013). In this study, the incidence of polyploidy has frequently been reported in Araceae Juss., Hydrocharitaceae Juss, Juncaginaceae Rich., Amaryllidaceae J. St.-Hil., Asparagaceae Juss., Dioscoreaceae R. Br., Liliaceae Juss., Melanthiaceae Batsch ex Borkh., Smilacaceae Vent. (Appendix 1). Analyses of ploidy levels distribution among these groups indicated that diploids (ca. 44%) and tetraploids (ca. 14%) prevail, with triploids (ca. 6%), pentaploids (ca. 2%), and hexaploids (ca. 4%) being found less frequently (Fig. 1, Appendix 1). Polyploidy is less frequent in Orchidaceae than in other families of Korean non-Commelinids monocots (Appendix 1), in agreement with previous reports for this region (Goldblatt, 1980; Ko et al., 2009; Rice et al., 2015, references therein). Despite the relatively high incidence of polyploidy in Korean non-Commelinids monocot flora and ease of inferring more recent polyploidy events based purely on increase of chromosome numbers, the clear inference of the mode of polyploids origin and inferences of the patterns of their post-polyploidization genome evolution are non-trivial and thus are not attempted here. These require rigorous phylogenetic analyses of the genera harboring polyploids to infer putative parental species and subsequent molecular cytogenetic analyses as well as genome size measurements to infer the patterns of their genome evolution. Such data are available only for a handful of selected monocot taxa (Appendix 1) and thus, more indepth and group-oriented molecular cytological analyses are required to assist and guide species delimitation and interpretation of phylogenetic relationships and evolutionary patterns among Korean monocots (Choi et al., 2008; Jang et al., 2013; Jang and Weiss-Schnneeweiss, 2015).
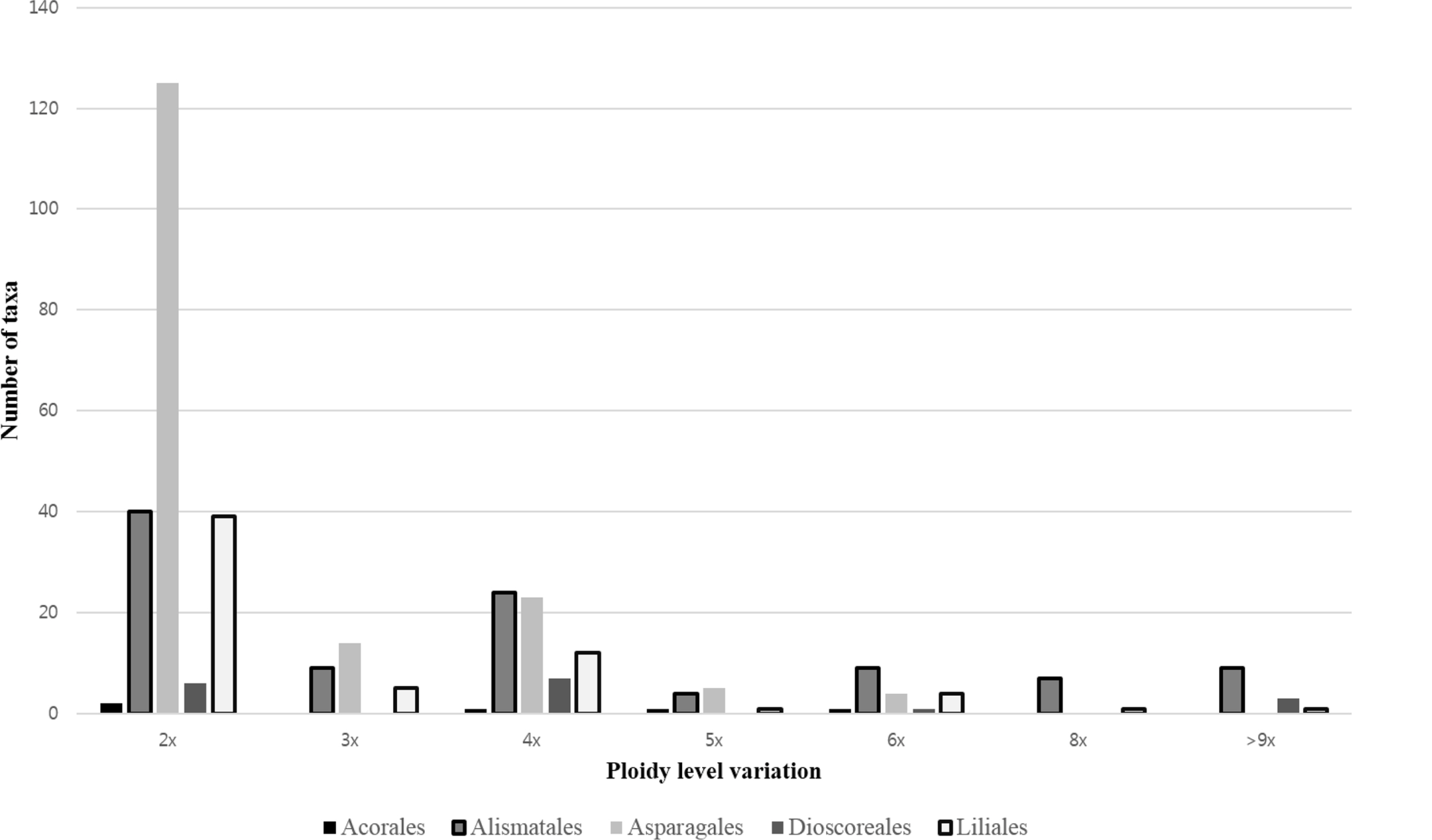
Distribution of ploidy level variation containing two to eight ploidy levels in non-Commelinids monocot species occurring in Korea (representing their worldwide distribution).
Genome size variation in non-commelinids monocots species native to Korea (in worldwide context)
The dynamics of genome size variation in a group of related diploid taxa can be very high despite lack of change in chromosome number. Genome size increase is, however, directly correlated to polyploidization, particularly recent one. Genome size changes in the absence of chromosome number changes are attributed to differential accumulation of various types of repetitive DNA elements (Leitch and Leitch, 2013). The range of genome sizes of Korean monocots falls within that reported in the Plant DNA C-values database which ranges from 0.061 pg/1C of DNA in Genlisea tuberosa Rivadavia, Gonella & A. Fleischm. (Fleischmann et al., 2014) to 152.33 pg/1C of DNA in Paris japonica Franch. (Pellicer et al., 2010). The 1C-values of species studied here differ nearly 150-fold and range from 0.3 pg in Spirodela polyrrhiza (L.) Schleid. (Araceae) to 44.5 pg in Trillium kamtschaticum Pall. ex Pursh (Melanthiaceae) (Fig. 2, Appendix 1). In general, the broad range of variation of genome sizes in flowering plants correlates with the differences of total karyotype length and incidence of polyploidy, but also correlates with other factors, like the life cycle types (annual/perennial) (Bennett, 1972; Chumová et al., 2015).

Distribution of genome size variation in non-Commelinids monocot species occurring in Korea (representing their worldwide distribution).
Patterns of genome evolution: the use of molecular cytogenetics and phylogenetic analyses in Plant Systematics
Extensive studies of chromosome numbers (including polyploidy incidence) and genome sizes in evolutionary context, aiming to elucidate the genome dynamics and often aiding taxonomic classifications have often been carried out in plants of agricultural importance or in model plants (Gong et al., 2012; Renny-Byfield et al., 2013; Novák et al., 2014; Zhang et al., 2014). However, recent advances in the advent of NGS technologies that enable large amounts of DNA sequence data to be generated in a single sequencing run at low cost, wild plants groups are now also amenable for in-depth genomic analyses. Such studies often address the evolution of polyploid complexes and focus on genome evolution in comparative context (e.g., polyploid and its lower-ploidy parental taxa) (Table 1) (Novák et al., 2010; Dodsworth et al., 2015; Weiss-Schneeweiss et al., 2015; McCann et al., 2018). These approaches allow for rapid identification of numerous types of DNA repeats providing new chromosomal markers that can be used in molecular cytological analyses applying in situ hybridization (fluorescence and genomic in situ hybridization; FISH and GISH, respectively) and thus, contributing to better understanding of the evolution of plant genomes (Table 1) (Renny-Byfield et al., 2010; Emadzade et al., 2014; Novák et al., 2014; Zhang et al., 2014; Jang and Weiss-Schneeweiss, 2015). Repetitive DNA fraction of plant genomes is composed of tandem repeats encompassing satellite DNAs, microsatellites and rDNAs (5S and 35S ribosomal RNA genes) as well as dispersed repeats represented by mobile genetic elements, known also as transposable elements. The latter comprise class I retroelements and class II DNA transposons (Weiss-Schneeweiss et al., 2015). In-depth analyses of repeatomes have recently been demonstrated to be informative for inferences of phylogenetic relationships in plants (Table 1) (Dodsworth et al., 2015, 2017; McCann et al., 2018).
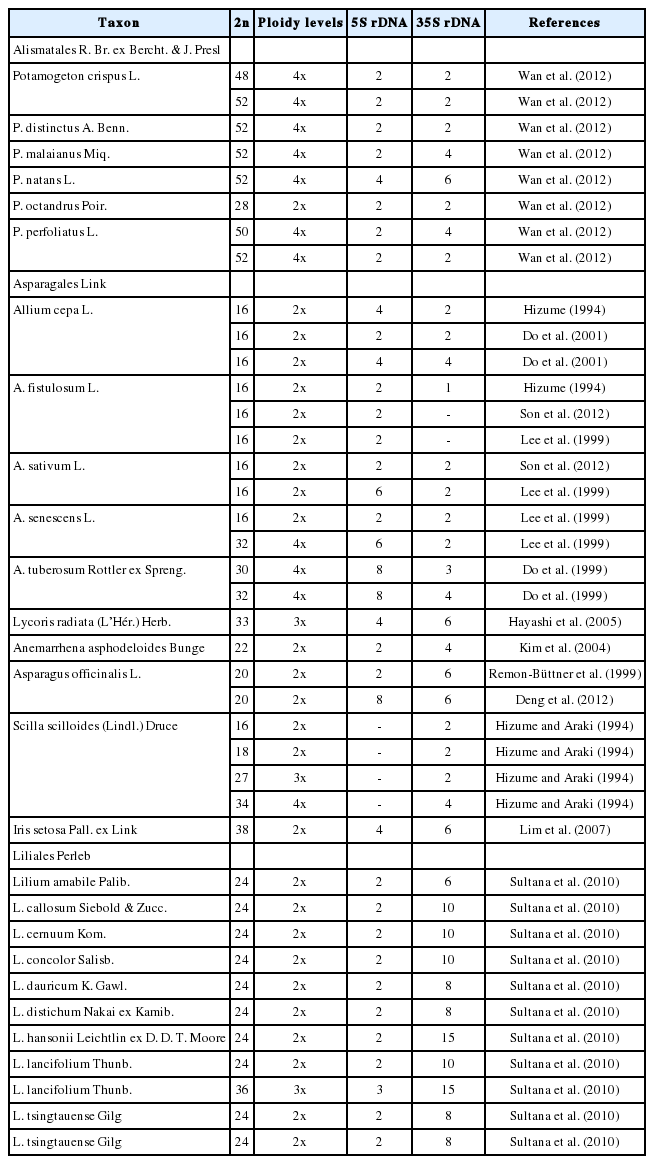
Summary of the chromosome numbers, ploidy level variation, and numbers of 5S and 35S rDNA signals in non-Commelinids monocot species occurring in Korea (representing their worldwide distribution)
Molecular cytogenetic mapping of the nuclear ribosomal RNA genes encoding for 35S (18S-5.8S-25S) and 5S rDNAs have proved useful for identifying the patterns and dynamics of chromosomal changes in closely related species groups (Jang et al., 2013, 2016a; Vitales et al., 2017). The distribution of rDNA loci has been reported for some Korean monocots, as summarized in Table 1 (data retrieved from Plant rDNA Database; http://www.plantrdnadatabase.com/, 2018 May 22). The number and localization of rDNA loci in diploids and polyploids was intensively studied in selected genera of Alismatales (Wan et al., 2012), Asparagales (Hizume, 1994; Hizume and Araki, 1994; Lee et al., 1999; Do et al., 1999, 2001; Remon-Büttner et al., 1999; Kim et al., 2004; Hayashi et al., 2005; Lim et al., 2007; Deng et al., 2012; Son et al., 2012), and Liliales (Sultana et al., 2010). A survey of rDNA loci numbers reported for Korean monocots indicated that rDNA loci number can vary at the interspecific level in the genera Allium, Lilium, and Potamogeton (between 2 and 6) (Table 1) regardless of chromosome number and ploidy level variation between species, as show for many other plant groups (Table 1, Appendix 1). The rDNA loci number variation within species or among closely related taxa have often been shown to be correlated with geographic and/or populational factors (e.g., Jang et al., 2016a). Thus, the localization of rDNA loci analyzed in comparative context aids not only the analyses of chromosomal structural changes, but when interpreted in phylogenetic context (e.g., Jang et al., 2013, 2016b), it also allows broader conclusions with implications for taxonomy. Monocot genomes are often more dynamically evolving than those of the dicots. Thus, further cytogenetic analyses of selected groups of Korean monocots will be undertaken to shed light into their genome evolution and evolutionary relationships. Such analyses should and will certainly include also populations and relatives from other geographical areas to allow for more robust conclusions to be drawn.
Acknowledgements
This work was supported by grants from the National Research Foundation of Korea (NRF) funded by the Korea government (grant numbers NRF-2018R1C1B6003170) to T.- S. Jang.
Notes
Conflict of Interest
The authors declare that there are no conflicts of interest.
